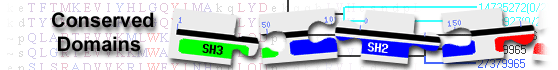|
Name |
Accession |
Description |
Interval |
E-value |
| guaA |
PRK00074 |
GMP synthase; Reviewed |
1-510 |
0e+00 |
|
GMP synthase; Reviewed
Pssm-ID: 234614 [Multi-domain] Cd Length: 511 Bit Score: 1045.40 E-value: 0e+00
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 1 MERELVIVVDFGGQYNQLIARRVRENNVYCEIVPYTYSVDKIKEKNPRGIIFTGGPNSVYDDNAPKISEDIFEIGVPVLG 80
Cdd:PRK00074 1 IHHDKILILDFGSQYTQLIARRVRELGVYSEIVPYDISAEEIRAFNPKGIILSGGPASVYEEGAPRADPEIFELGVPVLG 80
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 81 ICYGHQLICTTLGGKVESAQVREYGKTDVVLNNSSGLFSGIDKNESCWMSHTDFVSYPPEGFKIIGKSGESPVAAVENID 160
Cdd:PRK00074 81 ICYGMQLMAHQLGGKVERAGKREYGRAELEVDNDSPLFKGLPEEQDVWMSHGDKVTELPEGFKVIASTENCPIAAIANEE 160
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 161 KKIYGVQFHPEVEHTPFGKKMLSNFLFDICNLKGDWSMSSFVDEKIKSIKEEVGDKKVICAMSGGVDSSVAAMIVHKAVG 240
Cdd:PRK00074 161 RKFYGVQFHPEVTHTPQGKKLLENFVFDICGCKGDWTMENFIEEAIEEIREQVGDKKVILGLSGGVDSSVAAVLLHKAIG 240
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 241 KQLTCIFVDHGLLRKDEGDQVEDIFKKQFNMNFIRVNAEKRFLQKLKDISDPEKKRKIIGEEFIRVFEEEAKKLGEIAFL 320
Cdd:PRK00074 241 DQLTCVFVDHGLLRKNEAEQVMEMFREHFGLNLIHVDASDRFLSALAGVTDPEEKRKIIGREFIEVFEEEAKKLGGVKFL 320
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 321 VQGTIYPDVVESGLGT-SATIKSHHNVGGLPEDMDFKLIEPLRELFKDEVRAVGEELGIPHKLVWRQPFPGPGLAIRVLG 399
Cdd:PRK00074 321 AQGTLYPDVIESGGTKkAATIKSHHNVGGLPEDMKLKLVEPLRELFKDEVRKLGLELGLPEEIVYRHPFPGPGLAIRILG 400
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 400 NITEEKLQITRDADAIFREEIAKANLDETIWQYFACLPNIRSVGVMGDERTYCYTIALRAVISTDAMTSDWARIPYEVLD 479
Cdd:PRK00074 401 EVTKEKLDILREADAIFIEELRKAGLYDKIWQAFAVLLPVKSVGVMGDGRTYDYVVALRAVTSIDGMTADWARLPYDFLE 480
|
490 500 510
....*....|....*....|....*....|.
gi 219567478 480 KVSRRIVNEVKGVNRIVYDITSKPPATIEWE 510
Cdd:PRK00074 481 KISNRIINEVKGVNRVVYDITSKPPATIEWE 511
|
|
| GuaA2 |
COG0519 |
GMP synthase, PP-ATPase domain/subunit [Nucleotide transport and metabolism]; GMP synthase, ... |
1-510 |
0e+00 |
|
GMP synthase, PP-ATPase domain/subunit [Nucleotide transport and metabolism]; GMP synthase, PP-ATPase domain/subunit is part of the Pathway/BioSystem: Purine biosynthesis
Pssm-ID: 440285 [Multi-domain] Cd Length: 512 Bit Score: 954.66 E-value: 0e+00
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 1 MERELVIVVDFGGQYNQLIARRVRENNVYCEIVPYTYSVDKIKEKNPRGIIFTGGPNSVYDDNAPKISEDIFEIGVPVLG 80
Cdd:COG0519 1 MDKEIIIVLDFGGQYQQLIARRRREEGVYSEIIPPDTAAEEIKAKGPPGIILSGGPSSVYEEGAPPDDPELFELGGPILG 80
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 81 ICYGHQLICTTLGGKVESAQVREYGKTDVVLNNSSGLFSGIDKNESCWMSHTDFVSYPPEGFKIIGKSGESPVAAVENID 160
Cdd:COG0519 81 ICYGGQLMLHLLGGGVVRAERREYGGALLLVDDESDLFAGGPEELQVWMSHGDRVTELPPGFEVIASTENCPVAAIANEE 160
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 161 KKIYGVQFHPEVEHTPFGKKMLSNFLFDICNLKGDWSMSSFVDEKIKSIKEEVGDKKVICAMSGGVDSSVAAMIVHKAVG 240
Cdd:COG0519 161 RKLYGVQFHPEVVHTEQGKEILKNFLFDICGCKGDWTMENFIEEAIEEIREQVGDGKVICALSGGVDSSVAAALLHKAIG 240
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 241 KQLTCIFVDHGLLRKDEGDQVEDIFKKQFNMNFIRVNAEKRFLQKLKDISDPEKKRKIIGEEFIRVFEEEAKKLGEIAFL 320
Cdd:COG0519 241 DQLTCVFVDHGLLRKGEAEQVEETFKEHFGLNLIYVDASERFLSALKGVTDPEEKRKIIGEEFIEVFEEEAKKLGGAKFL 320
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 321 VQGTIYPDVVESG--LGTSATIKSHHNVGGLPEDMDFKLIEPLRELFKDEVRAVGEELGIPHKLVWRQPFPGPGLAIRVL 398
Cdd:COG0519 321 AQGTLYPDVIESGsvKGPAATIKSHHNVGGLPEDMKFKLVEPLRELFKDEVRALGRELGLPEEIVYRHPFPGPGLAIRIL 400
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 399 GNITEEKLQITRDADAIFREEIAKANLDETIWQYFACLPNIRSVGVMGDERTYCYTIALRAVISTDAMTSDWARIPYEVL 478
Cdd:COG0519 401 GEVTKEKLEILREADAIFIEELRKAGLYDKVWQAFAVLLPVKSVGVMGDERTYEYVVALRAVTSVDGMTADWARLPYEVL 480
|
490 500 510
....*....|....*....|....*....|..
gi 219567478 479 DKVSRRIVNEVKGVNRIVYDITSKPPATIEWE 510
Cdd:COG0519 481 ERISNRIINEVKGVNRVVYDITSKPPATIEWE 512
|
|
| PLN02347 |
PLN02347 |
GMP synthetase |
5-510 |
0e+00 |
|
GMP synthetase
Pssm-ID: 215197 [Multi-domain] Cd Length: 536 Bit Score: 709.53 E-value: 0e+00
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 5 LVIVVDFGGQYNQLIARRVRENNVYCEIVPYTYSVDKIKEKNPRGIIFTGGPNSVYDDNAPKISEDIFEI----GVPVLG 80
Cdd:PLN02347 12 VVLILDYGSQYTHLITRRVRELGVYSLLLSGTASLDRIASLNPRVVILSGGPHSVHVEGAPTVPEGFFDYcrerGVPVLG 91
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 81 ICYGHQLICTTLGGKVESAQVREYGKTDVVLNNSSGLFSGID--KNESCWMSHTDFVSYPPEGFKIIGKSGESPVAAVEN 158
Cdd:PLN02347 92 ICYGMQLIVQKLGGEVKPGEKQEYGRMEIRVVCGSQLFGDLPsgETQTVWMSHGDEAVKLPEGFEVVAKSVQGAVVAIEN 171
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 159 IDKKIYGVQFHPEVEHTPFGKKMLSNFLFDICNLKGDWSMSSFVDEKIKSIKEEVG-DKKVICAMSGGVDSSVAAMIVHK 237
Cdd:PLN02347 172 RERRIYGLQYHPEVTHSPKGMETLRHFLFDVCGVTADWKMQDVLEEQIELIKATVGpDEHVICALSGGVDSTVAATLVHK 251
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 238 AVGKQLTCIFVDHGLLRKDEGDQVEDIFKKQFNMNFIRVNAEKRFLQKLKDISDPEKKRKIIGEEFIRVFEEEA----KK 313
Cdd:PLN02347 252 AIGDRLHCVFVDNGLLRYKEQERVMETFKRDLHLPVTCVDASERFLSKLKGVTDPEKKRKIIGAEFIEVFDEFAhkleQK 331
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 314 LGEI-AFLVQGTIYPDVVES------GLGTSATIKSHHNVGGLPEDMDFKLIEPLRELFKDEVRAVGEELGIPHKLVWRQ 386
Cdd:PLN02347 332 LGKKpAFLVQGTLYPDVIEScpppgsGRTHSHTIKSHHNVGGLPKDMKLKLIEPLKLLFKDEVRKLGRLLGVPEAFLKRH 411
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 387 PFPGPGLAIRVLGNITEE-KLQITRDADAIFREEIAKANLDETIWQYFACLPNIRSVGVMGDERTYCYTIALRAVISTDA 465
Cdd:PLN02347 412 PFPGPGLAVRVLGDVTEGnALDILRQVDEIFINSIKDAGLYDEIWQAFAVFLPVKSVGVQGDQRTHSHVVALRAVTSEDG 491
|
490 500 510 520
....*....|....*....|....*....|....*....|....*
gi 219567478 466 MTSDWARIPYEVLDKVSRRIVNEVKGVNRIVYDITSKPPATIEWE 510
Cdd:PLN02347 492 MTADWYHFEHKFLDDVSRKICNEVRGVNRVVYDITSKPPSTIEWE 536
|
|
| guaA_Cterm |
TIGR00884 |
GMP synthase (glutamine-hydrolyzing), C-terminal domain or B subunit; This protein of purine ... |
201-510 |
0e+00 |
|
GMP synthase (glutamine-hydrolyzing), C-terminal domain or B subunit; This protein of purine de novo biosynthesis is well-conserved. However, it appears to split into two separate polypeptide chains in most of the Archaea. This C-terminal region would be the larger subunit [Purines, pyrimidines, nucleosides, and nucleotides, Purine ribonucleotide biosynthesis]
Pssm-ID: 273321 [Multi-domain] Cd Length: 311 Bit Score: 551.95 E-value: 0e+00
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 201 FVDEKIKSIKEEVGDKKVICAMSGGVDSSVAAMIVHKAVGKQLTCIFVDHGLLRKDEGDQVEDIFKKQFNMNFIRVNAEK 280
Cdd:TIGR00884 2 FIEEAVEEIREQVGDAKVIIALSGGVDSSVAAVLAHRAIGDRLTCVFVDHGLLRKGEAEQVVKTFGDRLGLNLVYVDAKE 81
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 281 RFLQKLKDISDPEKKRKIIGEEFIRVFEEEAKKLGEIAFLVQGTIYPDVVESGLGTSATIKSHHNVGGLPEDMDFKLIEP 360
Cdd:TIGR00884 82 RFLSALKGVTDPEEKRKIIGRVFIEVFEREAKKIGDAEYLAQGTIYPDVIESAAGTAHVIKSHHNVGGLPEDMKLKLVEP 161
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 361 LRELFKDEVRAVGEELGIPHKLVWRQPFPGPGLAIRVLGNITEEKLQITRDADAIFREEIAKANLDETIWQYFACLPNIR 440
Cdd:TIGR00884 162 LRELFKDEVRKLGKELGLPEEIVWRHPFPGPGLAVRVLGEVTKEKLEILRRADAIVIEELKKAGLYDKVWQAFAVLLPVK 241
|
250 260 270 280 290 300 310
....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 441 SVGVMGDERTYCYTIALRAVISTDAMTSDWARIPYEVLDKVSRRIVNEVKGVNRIVYDITSKPPATIEWE 510
Cdd:TIGR00884 242 SVGVMGDGRTYGYVIALRAVESIDGMTADWARLPYDFLERISNRITNEVPGVNRVVYDITSKPPATIEWE 311
|
|
| GMP_synthase_C |
cd01997 |
C-terminal domain of GMP synthetase (glutamine-hydrolyzing); The C-terminal domain of GMP ... |
209-510 |
0e+00 |
|
C-terminal domain of GMP synthetase (glutamine-hydrolyzing); The C-terminal domain of GMP synthetase (glutamine-hydrolyzing, EC 6.3.5.2) contains two subdomains: the ATP pyrophosphatase domain at the N-terminal and the dimerization domain at the C-terminal end. The ATP-PPase domain is a twisted, five-stranded parallel beta-sheet sandwiched between helical layers. It has a signature nucleotide-binding motif, or PP-loop, at the end of the first-beta strand. GMP synthetase is a homodimer, but in some archaea, it is a heterodimer made up of ATP pyrophosphatase (ATP-PPase) and a GATase subunit. In eukaryotes and bacteria, the two catalytic units are encoded by a single gene producing a two-domain-type GMP with a GATase domain in the N-terminus and an ATP-PPase domain in the C-terminus.
Pssm-ID: 467501 [Multi-domain] Cd Length: 311 Bit Score: 520.95 E-value: 0e+00
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 209 IKEEVGDKKVICAMSGGVDSSVAAMIVHKAVGK-QLTCIFVDHGLLRKDEGDQVEDIFKKQFNMNFIRVNAEKRFLQKLK 287
Cdd:cd01997 1 IKRTVGDKKVLCLVSGGVDSTVCAALLHKALGDeRVIAVHIDNGLMRKNESEQVEEALKKLGVINLAKVDASKRFLKKLK 80
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 288 DISDPEKKRKIIGEEFIRVFEEEAKKLG---EIAFLVQGTIYPDVVESG----LGTSATIKSHHNVGGLPED-MDFKLIE 359
Cdd:cd01997 81 GVTDPEEKRKIIGDTFIEVFDEVAKELNldpDDVYLAQGTLYPDLIESAsslaSSKADTIKTHHNVGGLPRElLKGKLVE 160
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 360 PLRELFKDEVRAVGEELGIPHKLVWRQPFPGPGLAIRVLGNITEEKLQITRDADAIFREEIAKANLDETIWQYFACLPNI 439
Cdd:cd01997 161 PLRDLFKDEVRELGRELGLPEELVWRHPFPGPGLAIRVLGEVTPEKLEILREADAIVEEELREAGLYDKISQAFAVLLPI 240
|
250 260 270 280 290 300 310
....*....|....*....|....*....|....*....|....*....|....*....|....*....|.
gi 219567478 440 RSVGVMGDERTYCYTIALRAVISTDAMTSDWARIPYEVLDKVSRRIVNEVKGVNRIVYDITSKPPATIEWE 510
Cdd:cd01997 241 KSVGVQGDGRTYGYVVALRAVETEDFMTAEWARPPYEVLDKISNRITNEVPGVNRVVYDITSKPPATIEWE 311
|
|
| GATase1_GMP_Synthase |
cd01742 |
Type 1 glutamine amidotransferase (GATase1) domain found in GMP synthetase; Type 1 glutamine ... |
6-186 |
3.42e-102 |
|
Type 1 glutamine amidotransferase (GATase1) domain found in GMP synthetase; Type 1 glutamine amidotransferase (GATase1) domain found in GMP synthetase. GMP synthetase is a glutamine amidotransferase from the de novo purine biosynthetic pathway. Glutamine amidotransferase (GATase) activity catalyse the transfer of ammonia from the amide side chain of glutamine to an acceptor substrate. GMP synthetase catalyses the amination of the nucleotide precursor xanthosine 5'-monophosphate to form GMP. GMP synthetase belongs to the triad family of amidotransferases having a conserved Cys-His-Glu catalytic triad in the glutaminase active site.
Pssm-ID: 153213 [Multi-domain] Cd Length: 181 Bit Score: 304.07 E-value: 3.42e-102
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 6 VIVVDFGGQYNQLIARRVRENNVYCEIVPYTYSVDKIKEKNPRGIIFTGGPNSVYDDNAPKISEDIFEIGVPVLGICYGH 85
Cdd:cd01742 1 ILILDFGSQYTHLIARRVRELGVYSEILPNTTPLEEIKLKNPKGIILSGGPSSVYEEDAPRVDPEIFELGVPVLGICYGM 80
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 86 QLICTTLGGKVESAQVREYGKTDVVLNNSSGLFSGIDKNESCWMSHTDFVSYPPEGFKIIGKSGESPVAAVENIDKKIYG 165
Cdd:cd01742 81 QLIAKALGGKVERGDKREYGKAEIEIDDSSPLFEGLPDEQTVWMSHGDEVVKLPEGFKVIASSDNCPVAAIANEEKKIYG 160
|
170 180
....*....|....*....|.
gi 219567478 166 VQFHPEVEHTPFGKKMLSNFL 186
Cdd:cd01742 161 VQFHPEVTHTEKGKEILKNFL 181
|
|
| guaA_Nterm |
TIGR00888 |
GMP synthase (glutamine-hydrolyzing), N-terminal domain or A subunit; This protein of purine ... |
6-193 |
1.75e-93 |
|
GMP synthase (glutamine-hydrolyzing), N-terminal domain or A subunit; This protein of purine de novo biosynthesis is well-conserved. However, it appears to split into two separate polypeptide chains in most of the Archaea. This N-terminal region would be the smaller subunit. [Purines, pyrimidines, nucleosides, and nucleotides, Purine ribonucleotide biosynthesis]
Pssm-ID: 129966 [Multi-domain] Cd Length: 188 Bit Score: 282.28 E-value: 1.75e-93
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 6 VIVVDFGGQYNQLIARRVRENNVYCEIVPYTYSVDKIKEKNPRGIIFTGGPNSVYDDNAPKISEDIFEIGVPVLGICYGH 85
Cdd:TIGR00888 1 ILVLDFGSQYTQLIARRLRELGVYSELVPNTTPLEEIREKNPKGIILSGGPSSVYAENAPRADEKIFELGVPVLGICYGM 80
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 86 QLICTTLGGKVESAQVREYGKTDVVLNNSSGLFSGIDKNESCWMSHTDFVSYPPEGFKIIGKSGESPVAAVENIDKKIYG 165
Cdd:TIGR00888 81 QLMAKQLGGEVGRAEKREYGKAELEILDEDDLFRGLPDESTVWMSHGDKVKELPEGFKVLATSDNCPVAAMAHEEKPIYG 160
|
170 180
....*....|....*....|....*...
gi 219567478 166 VQFHPEVEHTPFGKKMLSNFLFDICNLK 193
Cdd:TIGR00888 161 VQFHPEVTHTEYGNELLENFVYDVCGCE 188
|
|
| GuaA1 |
COG0518 |
GMP synthase, glutamine amidotransferase domain [Nucleotide transport and metabolism]; GMP ... |
6-189 |
1.02e-69 |
|
GMP synthase, glutamine amidotransferase domain [Nucleotide transport and metabolism]; GMP synthase, glutamine amidotransferase domain is part of the Pathway/BioSystem: Purine biosynthesis
Pssm-ID: 440284 [Multi-domain] Cd Length: 225 Bit Score: 222.51 E-value: 1.02e-69
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 6 VIVVD---FGGQYNQLIARRVREN-------NVY-CEIVPYTYSVDkikekNPRGIIFTGGPNSVYDD-----NAPKISE 69
Cdd:COG0518 2 ILILDhdpFGGQYPGLIARRLREAgieldvlRVYaGEILPYDPDLE-----DPDGLILSGGPMSVYDEdpwleDEPALIR 76
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 70 DIFEIGVPVLGICYGHQLICTTLGGKVESAQVREYGKTDVVLNNSSGLFSGIDKNESCWMSHTDFVSYPPEGFKIIGKSG 149
Cdd:COG0518 77 EAFELGKPVLGICYGAQLLAHALGGKVEPGPGREIGWAPVELTEADPLFAGLPDEFTVWMSHGDTVTELPEGAEVLASSD 156
|
170 180 190 200 210 220 230
....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 150 ESPVAAVEnIDKKIYGVQFHPEVEHT------------------------------PFGKKMLSNFLFDI 189
Cdd:COG0518 157 NCPNQAFR-YGRRVYGVQFHPEVTHTmmeawleeradelaaeellaeaslhdpelrEAGRRLLRNFLREI 225
|
|
| GMP_synt_C |
pfam00958 |
GMP synthase C terminal domain; GMP synthetase is a glutamine amidotransferase from the de ... |
418-509 |
1.01e-64 |
|
GMP synthase C terminal domain; GMP synthetase is a glutamine amidotransferase from the de novo purine biosynthetic pathway. This family is the C-terminal domain specific to the GMP synthases EC:6.3.5.2. In prokaryotes this domain mediates dimerization. Eukaryotic GMP synthases are monomers. This domain in eukaryotes includes several large insertions that may form globular domains.
Pssm-ID: 425963 [Multi-domain] Cd Length: 92 Bit Score: 204.57 E-value: 1.01e-64
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 418 EEIAKANLDETIWQYFACLPNIRSVGVMGDERTYCYTIALRAVISTDAMTSDWARIPYEVLDKVSRRIVNEVKGVNRIVY 497
Cdd:pfam00958 1 EEIKKAGLYRKIWQAFAVLLPVKSVGVMGDERTYEYVVALRAVTSTDGMTADWARLPYEVLEKISNRIVNEVPGVNRVVY 80
|
90
....*....|..
gi 219567478 498 DITSKPPATIEW 509
Cdd:pfam00958 81 DITSKPPATIEW 92
|
|
| PRK00758 |
PRK00758 |
GMP synthase subunit A; Validated |
6-190 |
1.14e-54 |
|
GMP synthase subunit A; Validated
Pssm-ID: 179112 [Multi-domain] Cd Length: 184 Bit Score: 181.59 E-value: 1.14e-54
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 6 VIVVDFGGQYNQLIARRVRENNVYCEIVPYTYSVDKIKEkNPRGIIFTGGPNSvydDNAPKISEDIFEIGVPVLGICYGH 85
Cdd:PRK00758 2 IVVVDNGGQYNHLIHRTLRYLGVDAKIIPNTTPVEEIKA-FEDGLILSGGPDI---ERAGNCPEYLKELDVPILGICLGH 77
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 86 QLICTTLGGKVESAQVREYGKTDVVLNNSSGLFSGIDKNESCWMSHTDFVSYPPEGFKIIGKSGESPVAAVENIDKKIYG 165
Cdd:PRK00758 78 QLIAKAFGGEVGRGEYGEYALVEVEILDEDDILKGLPPEIRVWASHADEVKELPDGFEILARSDICEVEAMKHKEKPIYG 157
|
170 180
....*....|....*....|....*
gi 219567478 166 VQFHPEVEHTPFGKKMLSNFLfDIC 190
Cdd:PRK00758 158 VQFHPEVAHTEYGEEIFKNFL-EIC 181
|
|
| GATase |
pfam00117 |
Glutamine amidotransferase class-I; |
7-189 |
4.89e-53 |
|
Glutamine amidotransferase class-I;
Pssm-ID: 395067 [Multi-domain] Cd Length: 188 Bit Score: 177.43 E-value: 4.89e-53
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 7 IVVDFGGQYNQLIARRVRENNVYCEIVPYTYSVDKIKEKNPRGIIFTGGPNSVYD-DNAPKISEDIFEIGVPVLGICYGH 85
Cdd:pfam00117 1 LLIDNGDSFTYNLARALRELGVEVTVVPNDTPAEEILEENPDGIILSGGPGSPGAaGGAIEAIREARELKIPILGICLGH 80
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 86 QLICTTLGGKVESAQVREY-GKTDVVLNNSSGLFSGIDKNESCWMSHTDFV--SYPPEGFKIIGKS--GESPVAAVENiD 160
Cdd:pfam00117 81 QLLALAFGGKVVKAKKFGHhGKNSPVGDDGCGLFYGLPNVFIVRRYHSYAVdpDTLPDGLEVTATSenDGTIMGIRHK-K 159
|
170 180
....*....|....*....|....*....
gi 219567478 161 KKIYGVQFHPEVEHTPFGKKMLSNFLFDI 189
Cdd:pfam00117 160 LPIFGVQFHPESILTPHGPEILFNFFIKA 188
|
|
| GATase1_1 |
cd01741 |
Subgroup of proteins having the Type 1 glutamine amidotransferase (GATase1) domain; This group ... |
49-186 |
4.28e-28 |
|
Subgroup of proteins having the Type 1 glutamine amidotransferase (GATase1) domain; This group contains a subgroup of proteins having the Type 1 glutamine amidotransferase (GATase1) domain. GATase activity catalyses the transfer of ammonia from the amide side chain of glutamine to an acceptor substrate. Glutamine amidotransferases (GATase) includes the triad family of amidotransferases which have a conserved Cys-His-Glu catalytic triad in the glutaminase active site. In this subgroup this triad is conserved. GATase activity can be found in a range of biosynthetic enzymes, including: glutamine amidotransferase, formylglycinamide ribonucleotide, GMP synthetase , anthranilate synthase component II, glutamine-dependent carbamoyl phosphate synthase, cytidine triphosphate synthetase, gamma-glutamyl hydrolase, imidazole glycerol phosphate synthase and, cobyric acid synthase. Glutamine amidotransferase (GATase) domains can occur either as single polypeptides, as in glutamine amidotransferases, or as domains in a much larger multifunctional synthase protein, such as CPSase.
Pssm-ID: 153212 [Multi-domain] Cd Length: 188 Bit Score: 110.41 E-value: 4.28e-28
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 49 GIIFTGGPNSVYDDNAPKISE------DIFEIGVPVLGICYGHQLICTTLGGKVE-SAQVREYGKTDVVLNN---SSGLF 118
Cdd:cd01741 49 GLVILGGPMSVDEDDYPWLKKlkelirQALAAGKPVLGICLGHQLLARALGGKVGrNPKGWEIGWFPVTLTEagkADPLF 128
|
90 100 110 120 130 140
....*....|....*....|....*....|....*....|....*....|....*....|....*...
gi 219567478 119 SGIDKNESCWMSHTDFVSYPPEGFKIIGKSGESPVAAVEnIDKKIYGVQFHPEvehtpfgKKMLSNFL 186
Cdd:cd01741 129 AGLPDEFPVFHWHGDTVVELPPGAVLLASSEACPNQAFR-YGDRALGLQFHPE-------ERLLRNFL 188
|
|
| PRK05670 |
PRK05670 |
anthranilate synthase component II; Provisional |
38-186 |
6.05e-26 |
|
anthranilate synthase component II; Provisional
Pssm-ID: 235552 [Multi-domain] Cd Length: 189 Bit Score: 104.44 E-value: 6.05e-26
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 38 SVDKIKEKNPRGIIFTGGPNSvyddnaPK---ISEDI---FEIGVPVLGICYGHQLICTTLGGKVESAQVREYGKTDVVL 111
Cdd:PRK05670 35 TLEEIEALNPDAIVLSPGPGT------PAeagISLELireFAGKVPILGVCLGHQAIGEAFGGKVVRAKEIMHGKTSPIE 108
|
90 100 110 120 130 140 150
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*..
gi 219567478 112 NNSSGLFSGIDKNESCWMSHTDFV--SYPPEGFKIIGKSGESPVAAVENIDKKIYGVQFHPEVEHTPFGKKMLSNFL 186
Cdd:PRK05670 109 HDGSGIFAGLPNPFTVTRYHSLVVdrESLPDCLEVTAWTDDGEIMGVRHKELPIYGVQFHPESILTEHGHKLLENFL 185
|
|
| GATase1_Anthranilate_Synthase |
cd01743 |
Type 1 glutamine amidotransferase (GATase1) domain found in Anthranilate synthase; Type 1 ... |
15-186 |
7.32e-26 |
|
Type 1 glutamine amidotransferase (GATase1) domain found in Anthranilate synthase; Type 1 glutamine amidotransferase (GATase1) domain found in Anthranilate synthase (ASase). This group contains proteins similar to para-aminobenzoate (PABA) synthase and ASase. These enzymes catalyze similar reactions and produce similar products, PABA and ortho-aminobenzoate (anthranilate). Each enzyme is composed of non-identical subunits: a glutamine amidotransferase subunit (component II) and a subunit that produces an aminobenzoate products (component I). ASase catalyses the synthesis of anthranilate from chorismate and glutamine and is a tetrameric protein comprising two copies each of components I and II. Component II of ASase belongs to the family of triad GTases which hydrolyze glutamine and transfer nascent ammonia between the active sites. In some bacteria, such as Escherichia coli, component II can be much larger than in other organisms, due to the presence of phosphoribosyl-anthranilate transferase (PRTase) activity. PRTase catalyses the second step in tryptophan biosynthesis and results in the addition of 5-phosphoribosyl-1-pyrophosphate to anthranilate to create N-5'-phosphoribosyl-anthranilate. In E.coli, the first step in the conversion of chorismate to PABA involves two proteins: PabA and PabB which co-operate to transfer the amide nitrogen of glutamine to chorismate forming 4-amino-4 deoxychorismate (ADC). PabA acts as a glutamine amidotransferase, supplying an amino group to PabB, which carries out the amination reaction. A third protein PabC then mediates elimination of pyruvate and aromatization to give PABA. Several organisms have bipartite proteins containing fused domains homologous to PabA and PabB commonly called PABA synthases. These hybrid PABA synthases may produce ADC and not PABA.
Pssm-ID: 153214 [Multi-domain] Cd Length: 184 Bit Score: 104.15 E-value: 7.32e-26
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 15 YNqlIARRVRENNVYCEIVPYT-YSVDKIKEKNPRGIIFTGGPNSvyddnaPKISEDIFEI------GVPVLGICYGHQL 87
Cdd:cd01743 12 YN--LVQYLRELGAEVVVVRNDeITLEELELLNPDAIVISPGPGH------PEDAGISLEIiralagKVPILGVCLGHQA 83
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 88 ICTTLGGKVESAQVREYGKTDVVLNNSSGLFSGIDKNESCWMSHT---DFVSyPPEGFKIIGKSGESPVAAVENIDKKIY 164
Cdd:cd01743 84 IAEAFGGKVVRAPEPMHGKTSEIHHDGSGLFKGLPQPFTVGRYHSlvvDPDP-LPDLLEVTASTEDGVIMALRHRDLPIY 162
|
170 180
....*....|....*....|..
gi 219567478 165 GVQFHPEVEHTPFGKKMLSNFL 186
Cdd:cd01743 163 GVQFHPESILTEYGLRLLENFL 184
|
|
| PabA |
COG0512 |
Anthranilate/para-aminobenzoate synthase component II (glutamine amidotransferase) [Amino acid ... |
15-186 |
5.35e-25 |
|
Anthranilate/para-aminobenzoate synthase component II (glutamine amidotransferase) [Amino acid transport and metabolism, Coenzyme transport and metabolism]; Anthranilate/para-aminobenzoate synthase component II (glutamine amidotransferase) is part of the Pathway/BioSystem: Aromatic amino acid biosynthesis
Pssm-ID: 440278 [Multi-domain] Cd Length: 189 Bit Score: 101.65 E-value: 5.35e-25
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 15 YNqlIARRVRENNVYCEIVPYT-YSVDKIKEKNPRGIIFTGGPNSVydDNAPkISEDI---FEIGVPVLGICYGHQLICT 90
Cdd:COG0512 12 YN--LVQYLGELGAEVVVVRNDeITLEEIEALAPDGIVLSPGPGTP--EEAG-ISLEViraFAGKIPILGVCLGHQAIGE 86
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 91 TLGGKVESAQVREYGKTDVVLNNSSGLFSGIDKNESCWMSHTDFVSYP--PEGFKIIGKSGESPVAAVENIDKKIYGVQF 168
Cdd:COG0512 87 AFGGKVVRAPEPMHGKTSPITHDGSGLFAGLPNPFTATRYHSLVVDREtlPDELEVTAWTEDGEIMGIRHRELPIEGVQF 166
|
170
....*....|....*...
gi 219567478 169 HPEVEHTPFGKKMLSNFL 186
Cdd:COG0512 167 HPESILTEHGHQLLANFL 184
|
|
| PRK14607 |
PRK14607 |
bifunctional anthranilate synthase component II/anthranilate phosphoribosyltransferase; |
38-186 |
2.83e-22 |
|
bifunctional anthranilate synthase component II/anthranilate phosphoribosyltransferase;
Pssm-ID: 237764 [Multi-domain] Cd Length: 534 Bit Score: 100.18 E-value: 2.83e-22
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 38 SVDKIKEKNPRGIIFTGGPNSvyddnaPK---ISEDI---FEIGVPVLGICYGHQLICTTLGGKVESAQVREYGKTDVVL 111
Cdd:PRK14607 36 TIEEIEALNPSHIVISPGPGR------PEeagISVEVirhFSGKVPILGVCLGHQAIGYAFGGKIVHAKRILHGKTSPID 109
|
90 100 110 120 130 140 150
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*..
gi 219567478 112 NNSSGLFSGIDKNESCWMSHTDFV--SYPPEGFKIIGKSGESPVAAVENIDKKIYGVQFHPEVEHTPFGKKMLSNFL 186
Cdd:PRK14607 110 HNGKGLFRGIPNPTVATRYHSLVVeeASLPECLEVTAKSDDGEIMGIRHKEHPIFGVQFHPESILTEEGKRILKNFL 186
|
|
| trpG |
CHL00101 |
anthranilate synthase component 2 |
38-186 |
1.90e-18 |
|
anthranilate synthase component 2
Pssm-ID: 214365 [Multi-domain] Cd Length: 190 Bit Score: 83.24 E-value: 1.90e-18
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 38 SVDKIKEKNPRGIIFTGGPNSVYDDNapkISEDI---FEIGVPVLGICYGHQLICTTLGGKVESAQVREYGKTDVVLNNS 114
Cdd:CHL00101 35 DLSKIKNLNIRHIIISPGPGHPRDSG---ISLDVissYAPYIPILGVCLGHQSIGYLFGGKIIKAPKPMHGKTSKIYHNH 111
|
90 100 110 120 130 140 150
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*
gi 219567478 115 SGLFSGIDKNESCWMSHTDFV--SYPPEGFKIIGKSGESPVAAVENID-KKIYGVQFHPEVEHTPFGKKMLSNFL 186
Cdd:CHL00101 112 DDLFQGLPNPFTATRYHSLIIdpLNLPSPLEITAWTEDGLIMACRHKKyKMLRGIQFHPESLLTTHGQQILRNFL 186
|
|
| PRK07649 |
PRK07649 |
aminodeoxychorismate/anthranilate synthase component II; |
38-186 |
3.42e-16 |
|
aminodeoxychorismate/anthranilate synthase component II;
Pssm-ID: 181066 [Multi-domain] Cd Length: 195 Bit Score: 76.77 E-value: 3.42e-16
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 38 SVDKIKEKNPRGIIFTGGPNSvydDNAPKISEDI---FEIGVPVLGICYGHQLICTTLGGKVESAQVREYGKTDVVLNNS 114
Cdd:PRK07649 35 TISDIENMKPDFLMISPGPCS---PNEAGISMEViryFAGKIPIFGVCLGHQSIAQVFGGEVVRAERLMHGKTSLMHHDG 111
|
90 100 110 120 130 140 150
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....
gi 219567478 115 SGLFSGIDKNESCWMSHTDFVSYP--PEGFKIIGKSGESPVAAVENIDKKIYGVQFHPEVEHTPFGKKMLSNFL 186
Cdd:PRK07649 112 KTIFSDIPNPFTATRYHSLIVKKEtlPDCLEVTSWTEEGEIMAIRHKTLPIEGVQFHPESIMTSHGKELLQNFI 185
|
|
| GATase1_CPSase |
cd01744 |
Small chain of the glutamine-dependent form of carbamoyl phosphate synthase, CPSase II; This ... |
6-171 |
1.53e-15 |
|
Small chain of the glutamine-dependent form of carbamoyl phosphate synthase, CPSase II; This group of sequences represents the small chain of the glutamine-dependent form of carbamoyl phosphate synthase, CPSase II. CPSase II catalyzes the production of carbomyl phosphate (CP) from bicarbonate, glutamine and two molecules of MgATP. The reaction is believed to proceed by a series of four biochemical reactions involving a minimum of three discrete highly reactive intermediates. The synthesis of CP is critical for the initiation of two separate biosynthetic pathways. In one CP is coupled to aspartate, its carbon and nitrogen nuclei ultimately incorporated into the aromatic moieties of pyrimidine nucleotides. In the second pathway CP is condensed with ornithine at the start of the urea cycle and is utilized for the detoxification of ammonia and biosynthesis of arginine. CPSases may be encoded by one or by several genes, depending on the species. The E.coli enzyme is a heterodimer consisting of two polypeptide chains referred to as the small and large subunit. Ammonia an intermediate during the biosynthesis of carbomyl phosphate produced by the hydrolysis of glutamine in the small subunit of the enzyme is delivered via a molecular tunnel between the remotely located carboxyphosphate active site in the large subunit. CPSase IIs belong to the triad family of amidotransferases having a conserved Cys-His-Glu catalytic triad in the glutaminase active site. This group also contains the sequence from the mammalian urea cycle form which has lost the active site Cys, resulting in an ammonia-dependent form, CPSase I.
Pssm-ID: 153215 [Multi-domain] Cd Length: 178 Bit Score: 74.46 E-value: 1.53e-15
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 6 VIVVDFGGQYNQLiaRRVRENNVYCEIVPYTYSVDKIKEKNPRGIIFTGGPNSVYD-DNAPKISEDIFEIGVPVLGICYG 84
Cdd:cd01744 1 VVVIDFGVKHNIL--RELLKRGCEVTVVPYNTDAEEILKLDPDGIFLSNGPGDPALlDEAIKTVRKLLGKKIPIFGICLG 78
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 85 HQLICTTLGGKVESAQVREYGKTDVVLNNSSGlfsgidkneSCWMS---HtdfvsyppeGFKIIGKSGESPVAA------ 155
Cdd:cd01744 79 HQLLALALGAKTYKMKFGHRGSNHPVKDLITG---------RVYITsqnH---------GYAVDPDSLPGGLEVthvnln 140
|
170 180
....*....|....*....|..
gi 219567478 156 ---VENI---DKKIYGVQFHPE 171
Cdd:cd01744 141 dgtVEGIrhkDLPVFSVQFHPE 162
|
|
| PRK09065 |
PRK09065 |
glutamine amidotransferase; Provisional |
48-171 |
1.70e-15 |
|
glutamine amidotransferase; Provisional
Pssm-ID: 181635 [Multi-domain] Cd Length: 237 Bit Score: 75.77 E-value: 1.70e-15
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 48 RGIIFTGGPNSVyDDNAP---KISEDIFE---IGVPVLGICYGHQLICTTLGGKV-ESAQVREYGKTDVVLNNSSG---L 117
Cdd:PRK09065 56 AGVIITGSWAMV-TDRLDwseRTADWLRQaaaAGMPLLGICYGHQLLAHALGGEVgYNPAGRESGTVTVELHPAAAddpL 134
|
90 100 110 120 130
....*....|....*....|....*....|....*....|....*....|....
gi 219567478 118 FSGIDKNESCWMSHTDFVSYPPEGFKIIGKSGESPVAAVEnIDKKIYGVQFHPE 171
Cdd:PRK09065 135 FAGLPAQFPAHLTHLQSVLRLPPGAVVLARSAQDPHQAFR-YGPHAWGVQFHPE 187
|
|
| PLN02335 |
PLN02335 |
anthranilate synthase |
30-186 |
2.61e-15 |
|
anthranilate synthase
Pssm-ID: 177969 [Multi-domain] Cd Length: 222 Bit Score: 75.22 E-value: 2.61e-15
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 30 CEIVPY---TYSVDKIKEKNPRGIIFTGGPNSVYDDNapkIS-EDIFEIG--VPVLGICYGHQLICTTLGGKVesaqVRE 103
Cdd:PLN02335 43 CHFEVYrndELTVEELKRKNPRGVLISPGPGTPQDSG---ISlQTVLELGplVPLFGVCMGLQCIGEAFGGKI----VRS 115
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 104 -----YGKTDVVLNNS---SGLFSGIDKNESCWMSHTDFV---SYPPEGFKIIGKSGESPVAAVEN-IDKKIYGVQFHPE 171
Cdd:PLN02335 116 pfgvmHGKSSPVHYDEkgeEGLFSGLPNPFTAGRYHSLVIekdTFPSDELEVTAWTEDGLIMAARHrKYKHIQGVQFHPE 195
|
170
....*....|....*
gi 219567478 172 VEHTPFGKKMLSNFL 186
Cdd:PLN02335 196 SIITTEGKTIVRNFI 210
|
|
| CPSaseIIsmall |
TIGR01368 |
carbamoyl-phosphate synthase, small subunit; This model represents the whole of the small ... |
3-188 |
1.80e-14 |
|
carbamoyl-phosphate synthase, small subunit; This model represents the whole of the small chain of the glutamine-dependent form (EC 6.3.5.5) of carbamoyl phosphate synthase, CPSase II. The C-terminal domain has glutamine amidotransferase activity. Note that the sequence from the mammalian urea cycle form has lost the active site Cys, resulting in an ammonia-dependent form, CPSase I (EC 6.3.4.16). CPSases of pyrimidine biosynthesis, arginine biosynthesis, and the urea cycle may be encoded by one or by several genes, depending on the species. [Purines, pyrimidines, nucleosides, and nucleotides, Pyrimidine ribonucleotide biosynthesis]
Pssm-ID: 273580 [Multi-domain] Cd Length: 357 Bit Score: 74.58 E-value: 1.80e-14
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 3 RELVIVVDFGGQYNqlIARRVRENNVYCEIVPYTYSVDKIKEKNPRGIIFTGGPN--SVYDDNAPKISEDIFEIgvPVLG 80
Cdd:TIGR01368 172 GKRVVVIDFGVKRN--ILRRLVKRGCEVTVVPYDTDAEEIKKYNPDGIFLSNGPGdpAAVEPAIETIRKLLEKI--PIFG 247
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 81 ICYGHQLICTTLGGKVESAQVREYGKTDVVLNNSSGlfsgidkneSCWMS---H---TDFVSYPPEGFKIIGKS-GESPV 153
Cdd:TIGR01368 248 ICLGHQLLALAFGAKTYKMKFGHRGGNHPVKDLITG---------RVEITsqnHgyaVDPDSLPAGDLEVTHVNlNDGTV 318
|
170 180 190
....*....|....*....|....*....|....*
gi 219567478 154 AAVENIDKKIYGVQFHPEVEHTPFGkkmlSNFLFD 188
Cdd:TIGR01368 319 EGIRHKDLPVFSVQYHPEASPGPHD----TEYLFD 349
|
|
| PRK08007 |
PRK08007 |
aminodeoxychorismate synthase component 2; |
38-186 |
2.16e-14 |
|
aminodeoxychorismate synthase component 2;
Pssm-ID: 181194 [Multi-domain] Cd Length: 187 Bit Score: 71.49 E-value: 2.16e-14
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 38 SVDKIKEKNPRGIIFTGGPNSvydDNAPKISEDI---FEIGVPVLGICYGHQLICTTLGGKVESAQVREYGKTDVVLNNS 114
Cdd:PRK08007 35 TLADIDALKPQKIVISPGPCT---PDEAGISLDVirhYAGRLPILGVCLGHQAMAQAFGGKVVRAAKVMHGKTSPITHNG 111
|
90 100 110 120 130 140 150
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....
gi 219567478 115 SGLFSGIDKNESCWMSHTDFVSYP--PEGFKIIGKSGESPVAAVENIDKKIYGVQFHPEVEHTPFGKKMLSNFL 186
Cdd:PRK08007 112 EGVFRGLANPLTVTRYHSLVVEPDslPACFEVTAWSETREIMGIRHRQWDLEGVQFHPESILSEQGHQLLANFL 185
|
|
| trpG_papA |
TIGR00566 |
glutamine amidotransferase of anthranilate synthase or aminodeoxychorismate synthase; This ... |
40-186 |
5.54e-14 |
|
glutamine amidotransferase of anthranilate synthase or aminodeoxychorismate synthase; This model describes the glutamine amidotransferase domain or peptide of the tryptophan-biosynthetic pathway enzyme anthranilate synthase or of the folate biosynthetic pathway enzyme para-aminobenzoate synthase. In at least one case, a single polypeptide from Bacillus subtilis was shown to have both functions. This model covers a subset of the sequences described by the Pfam model GATase.
Pssm-ID: 273144 [Multi-domain] Cd Length: 188 Bit Score: 70.20 E-value: 5.54e-14
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 40 DKIKEKNPRGIIFTGGPnsVYDDNApKISEDIFE--IG-VPVLGICYGHQLICTTLGGKVESAQVREYGKTDVVLNNSSG 116
Cdd:TIGR00566 37 QEIEALLPLLIVISPGP--CTPNEA-GISLEAIRhfAGkLPILGVCLGHQAMGQAFGGDVVRANTVMHGKTSEIEHNGAG 113
|
90 100 110 120 130 140 150
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*
gi 219567478 117 LFSGIDKNESCWMSHTDFVSypPEG----FKIIGKSGESP-VAAVENIDKKIYGVQFHPEVEHTPFGKKMLSNFL 186
Cdd:TIGR00566 114 IFRGLFNPLTATRYHSLVVE--PETlptcFPVTAWEEENIeIMAIRHRDLPLEGVQFHPESILSEQGHQLLANFL 186
|
|
| Peptidase_C26 |
pfam07722 |
Peptidase C26; These peptidases have gamma-glutamyl hydrolase activity; that is they catalyze ... |
49-171 |
4.40e-13 |
|
Peptidase C26; These peptidases have gamma-glutamyl hydrolase activity; that is they catalyze the cleavage of the gamma-glutamyl bond in poly-gamma-glutamyl substrates. They are structurally related to pfam00117, but contain extensions in four loops and at the C terminus.
Pssm-ID: 429620 [Multi-domain] Cd Length: 219 Bit Score: 68.44 E-value: 4.40e-13
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 49 GIIFTGGPN---SVYDDNA-PKISE-----DIFEI---------GVPVLGICYGHQLICTTLGG--------------KV 96
Cdd:pfam07722 61 GLLLTGGPNvdpHFYGEEPsESGGPydparDAYELaliraalarGKPILGICRGFQLLNVALGGtlyqdiqeqpgftdHR 140
|
90 100 110 120 130 140 150
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....
gi 219567478 97 ESAQVREYGKTDVVLNNSSGLFSGIDKNESCWMS--HTDFVSYPPEGFKIIGKSGESPVAAVE--NIDKKIYGVQFHPE 171
Cdd:pfam07722 141 EHCQVAPYAPSHAVNVEPGSLLASLLGSEEFRVNslHHQAIDRLAPGLRVEAVAPDGTIEAIEspNAKGFALGVQWHPE 219
|
|
| PRK07765 |
PRK07765 |
aminodeoxychorismate/anthranilate synthase component II; |
75-214 |
5.35e-13 |
|
aminodeoxychorismate/anthranilate synthase component II;
Pssm-ID: 181107 [Multi-domain] Cd Length: 214 Bit Score: 68.15 E-value: 5.35e-13
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 75 GVPVLGICYGHQLICTTLGGKVESAQVREYGKTDVVLNNSSGLFSGIDKNESCWMSHTDFV---SYPPEgFKIIGKSGES 151
Cdd:PRK07765 76 GTPLLGVCLGHQAIGVAFGATVDRAPELLHGKTSSVHHTGVGVLAGLPDPFTATRYHSLTIlpeTLPAE-LEVTARTDSG 154
|
90 100 110 120 130 140
....*....|....*....|....*....|....*....|....*....|....*....|...
gi 219567478 152 PVAAVENIDKKIYGVQFHPEVEHTPFGKKMLSNFLfDICNLKGDwsmssfvDEKIKSIKEEVG 214
Cdd:PRK07765 155 VIMAVRHRELPIHGVQFHPESVLTEGGHRMLANWL-TVCGWAPD-------EALVRRLENEVA 209
|
|
| PRK08857 |
PRK08857 |
aminodeoxychorismate/anthranilate synthase component II; |
39-186 |
2.64e-12 |
|
aminodeoxychorismate/anthranilate synthase component II;
Pssm-ID: 181566 [Multi-domain] Cd Length: 193 Bit Score: 65.67 E-value: 2.64e-12
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 39 VDKIKEKNPRGIIFTGGPNSVYDDNAPKISEDIFEIGVPVLGICYGHQLICTTLGGKVESAQVREYGKTDVVLNNSSGLF 118
Cdd:PRK08857 36 IDGIEALNPTHLVISPGPCTPNEAGISLQAIEHFAGKLPILGVCLGHQAIAQVFGGQVVRARQVMHGKTSPIRHTGRSVF 115
|
90 100 110 120 130 140 150
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*
gi 219567478 119 SGIDKNESCWMSHTDFVSYP--PEGFKIIG----KSGE-SPVAAVENIDKKIYGVQFHPEVEHTPFGKKMLSNFL 186
Cdd:PRK08857 116 KGLNNPLTVTRYHSLVVKNDtlPECFELTAwtelEDGSmDEIMGFQHKTLPIEAVQFHPESIKTEQGHQLLANFL 190
|
|
| CarA |
COG0505 |
Carbamoylphosphate synthase small subunit [Amino acid transport and metabolism, Nucleotide ... |
6-95 |
3.21e-12 |
|
Carbamoylphosphate synthase small subunit [Amino acid transport and metabolism, Nucleotide transport and metabolism]; Carbamoylphosphate synthase small subunit is part of the Pathway/BioSystem: Arginine biosynthesis
Pssm-ID: 440271 [Multi-domain] Cd Length: 361 Bit Score: 67.74 E-value: 3.21e-12
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 6 VIVVDFGGQYNqlIARRVRENNVYCEIVPYTYSVDKIKEKNPRGIIFTGGPN--SVYDDNAPKISEdIFEIGVPVLGICY 83
Cdd:COG0505 179 VVALDFGVKRN--ILRELAERGCRVTVVPATTSAEEILALNPDGVFLSNGPGdpAALDYAIETIRE-LLGKGIPIFGICL 255
|
90
....*....|..
gi 219567478 84 GHQLICTTLGGK 95
Cdd:COG0505 256 GHQLLALALGAK 267
|
|
| hisH |
PRK13181 |
imidazole glycerol phosphate synthase subunit HisH; Provisional |
73-186 |
3.23e-12 |
|
imidazole glycerol phosphate synthase subunit HisH; Provisional
Pssm-ID: 183878 [Multi-domain] Cd Length: 199 Bit Score: 65.66 E-value: 3.23e-12
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 73 EIGVPVLGICYGHQLICTT--------LG---GKV-----ESAQVREYGKTDVVLNNSSGLFSGIDKNESCWMSHTDFVs 136
Cdd:PRK13181 70 EKKQPVLGICLGMQLLFESseegnvkgLGlipGDVkrfrsEPLKVPQMGWNSVKPLKESPLFKGIEEGSYFYFVHSYYV- 148
|
90 100 110 120 130
....*....|....*....|....*....|....*....|....*....|...
gi 219567478 137 yPPEGFKIIGKS---GESPVAAVENidKKIYGVQFHPEVEHtPFGKKMLSNFL 186
Cdd:PRK13181 149 -PCEDPEDVLATteyGVPFCSAVAK--DNIYAVQFHPEKSG-KAGLKLLKNFA 197
|
|
| PRK06895 |
PRK06895 |
anthranilate synthase component II; |
78-186 |
1.74e-11 |
|
anthranilate synthase component II;
Pssm-ID: 235882 [Multi-domain] Cd Length: 190 Bit Score: 63.22 E-value: 1.74e-11
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 78 VLGICYGHQLICTTLGGKVES-AQVREYGKTDVVLNNSSGLFSGIDKNESCWMSHTDFVSYP--PEGFKIIGKSGESPVA 154
Cdd:PRK06895 75 ILGVCLGHQTLCEFFGGELYNlNNVRHGQQRPLKVRSNSPLFDGLPEEFNIGLYHSWAVSEEnfPTPLEITAVCDENVVM 154
|
90 100 110
....*....|....*....|....*....|..
gi 219567478 155 AVENIDKKIYGVQFHPEVEHTPFGKKMLSNFL 186
Cdd:PRK06895 155 AMQHKTLPIYGVQFHPESYISEFGEQILRNWL 186
|
|
| GATase1 |
cd01653 |
Type 1 glutamine amidotransferase (GATase1)-like domain; Type 1 glutamine amidotransferase ... |
6-105 |
1.78e-11 |
|
Type 1 glutamine amidotransferase (GATase1)-like domain; Type 1 glutamine amidotransferase (GATase1)-like domain. This group includes proteins similar to Class I glutamine amidotransferases, the intracellular PH1704 from Pyrococcus horikoshii, the C-terminal of the large catalase: Escherichia coli HP-II, Sinorhizobium meliloti Rm1021 ThuA. and, the A4 beta-galactosidase middle domain. The majority of proteins in this group have a reactive Cys found in the sharp turn between a beta strand and an alpha helix termed the nucleophile elbow. For Class I glutamine amidotransferases proteins which transfer ammonia from the amide side chain of glutamine to an acceptor substrate, this Cys forms a Cys-His-Glu catalytic triad in the active site. Glutamine amidotransferases activity can be found in a range of biosynthetic enzymes included in this cd: glutamine amidotransferase, formylglycinamide ribonucleotide, GMP synthetase, anthranilate synthase component II, glutamine-dependent carbamoyl phosphate synthase, cytidine triphosphate synthetase, gamma-glutamyl hydrolase, imidazole glycerol phosphate synthase and, cobyric acid synthase. For Pyrococcus horikoshii PH1704, the Cys of the nucleophile elbow together with a different His and, a Glu from an adjacent monomer form a catalytic triad different from the typical GATase1 triad. The E. coli HP-II C-terminal domain, S. meliloti Rm1021 ThuA and the A4 beta-galactosidase middle domain lack the catalytic triad typical GATaseI domains. GATase1-like domains can occur either as single polypeptides, as in Class I glutamine amidotransferases, or as domains in a much larger multifunctional synthase protein, such as CPSase.
Pssm-ID: 153210 [Multi-domain] Cd Length: 115 Bit Score: 61.08 E-value: 1.78e-11
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 6 VIVVDFGGQYN---QLIARRVRENNVYCEIVPYT--YSVDKIKEKNPRGIIFTGGPNSVYD--DNAPKISE--DIFEIGV 76
Cdd:cd01653 1 VAVLLFPGFEElelASPLDALREAGAEVDVVSPDggPVESDVDLDDYDGLILPGGPGTPDDlaRDEALLALlrEAAAAGK 80
|
90 100
....*....|....*....|....*....
gi 219567478 77 PVLGICYGHQLICTTLGGKVESAQVREYG 105
Cdd:cd01653 81 PILGICLGAQLLVLGVQFHPEAIDGAEAG 109
|
|
| PRK12564 |
PRK12564 |
carbamoyl-phosphate synthase small subunit; |
6-97 |
1.80e-11 |
|
carbamoyl-phosphate synthase small subunit;
Pssm-ID: 237139 [Multi-domain] Cd Length: 360 Bit Score: 65.48 E-value: 1.80e-11
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 6 VIVVDFGGQYNqlIARRVRENNVYCEIVPYTYSVDKIKEKNPRGIIFTGGPN--SVYDDNAPKISEdIFEIGVPVLGICY 83
Cdd:PRK12564 180 VVAIDFGVKRN--ILRELAERGCRVTVVPATTTAEEILALNPDGVFLSNGPGdpAALDYAIEMIRE-LLEKKIPIFGICL 256
|
90
....*....|....
gi 219567478 84 GHQLICTTLGGKVE 97
Cdd:PRK12564 257 GHQLLALALGAKTY 270
|
|
| PLN02771 |
PLN02771 |
carbamoyl-phosphate synthase (glutamine-hydrolyzing) |
6-116 |
2.04e-11 |
|
carbamoyl-phosphate synthase (glutamine-hydrolyzing)
Pssm-ID: 178370 [Multi-domain] Cd Length: 415 Bit Score: 65.77 E-value: 2.04e-11
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 6 VIVVDFGGQYNqlIARRVRENNVYCEIVPYTYSVDKIKEKNPRGIIFTGGPNsvyDDNA-PKISEDIFEI--GVPVLGIC 82
Cdd:PLN02771 243 VIAYDFGIKHN--ILRRLASYGCKITVVPSTWPASEALKMKPDGVLFSNGPG---DPSAvPYAVETVKELlgKVPVFGIC 317
|
90 100 110
....*....|....*....|....*....|....
gi 219567478 83 YGHQLICTTLGGKVESAQVREYGKTDVVLNNSSG 116
Cdd:PLN02771 318 MGHQLLGQALGGKTFKMKFGHHGGNHPVRNNRTG 351
|
|
| PRK12838 |
PRK12838 |
carbamoyl phosphate synthase small subunit; Reviewed |
6-171 |
9.04e-11 |
|
carbamoyl phosphate synthase small subunit; Reviewed
Pssm-ID: 183784 [Multi-domain] Cd Length: 354 Bit Score: 63.37 E-value: 9.04e-11
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 6 VIVVDFGgqYNQLIARRVRENNVYCEIVPYTYSVDKIKEKNPRGIIFTGGPNSV--YDDNAPKIsEDIFEiGVPVLGICY 83
Cdd:PRK12838 170 VALIDFG--YKKSILRSLSKRGCKVTVLPYDTSLEEIKNLNPDGIVLSNGPGDPkeLQPYLPEI-KKLIS-SYPILGICL 245
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 84 GHQLICTTLGGKVESAQVREYGKTDVVLNNSSGlfsgidkneSCWMS---HtDFV----SYPPEGFKI--IGKSGESpVA 154
Cdd:PRK12838 246 GHQLIALALGADTEKLPFGHRGANHPVIDLTTG---------RVWMTsqnH-GYVvdedSLDGTPLSVrfFNVNDGS-IE 314
|
170
....*....|....*..
gi 219567478 155 AVENIDKKIYGVQFHPE 171
Cdd:PRK12838 315 GLRHKKKPVLSVQFHPE 331
|
|
| PRK05665 |
PRK05665 |
amidotransferase; Provisional |
69-174 |
2.23e-10 |
|
amidotransferase; Provisional
Pssm-ID: 168162 [Multi-domain] Cd Length: 240 Bit Score: 60.98 E-value: 2.23e-10
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 69 EDIFEIGVPVLGICYGHQLICTTLGGKVESA-QVREYGKTDVVLNNSSGLFSGIDKNESCWMSHTDFVSYPPEGFKIIGK 147
Cdd:PRK05665 85 LKLYERGDKLLGVCFGHQLLALLLGGKAERAsQGWGVGIHRYQLAAHAPWMSPAVTELTLLISHQDQVTALPEGATVIAS 164
|
90 100
....*....|....*....|....*..
gi 219567478 148 SGESPVAAVeNIDKKIYGVQFHPEVEH 174
Cdd:PRK05665 165 SDFCPFAAY-HIGDQVLCFQGHPEFVH 190
|
|
| carA |
CHL00197 |
carbamoyl-phosphate synthase arginine-specific small subunit; Provisional |
6-188 |
2.78e-10 |
|
carbamoyl-phosphate synthase arginine-specific small subunit; Provisional
Pssm-ID: 214392 [Multi-domain] Cd Length: 382 Bit Score: 62.12 E-value: 2.78e-10
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 6 VIVVDFGGQYNqlIARRVRENNVYCEIVPYTYSVDKIKEKNPRGIIFTGGPN--SVYDDNAPKISEdIFEIGVPVLGICY 83
Cdd:CHL00197 195 IIVIDFGVKYN--ILRRLKSFGCSITVVPATSPYQDILSYQPDGILLSNGPGdpSAIHYGIKTVKK-LLKYNIPIFGICM 271
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 84 GHQLICTTLGGKVESAQVREYGktdvvLNNSSGLFSgidKNESCWMSH---TDFVSYPPEGFKIIGKS-GESPVAAVENI 159
Cdd:CHL00197 272 GHQILSLALEAKTFKLKFGHRG-----LNHPSGLNQ---QVEITSQNHgfaVNLESLAKNKFYITHFNlNDGTVAGISHS 343
|
170 180
....*....|....*....|....*....
gi 219567478 160 DKKIYGVQFHPEVEHTPFGkkmlSNFLFD 188
Cdd:CHL00197 344 PKPYFSVQYHPEASPGPHD----ADYLFE 368
|
|
| IMP_synth_hisH |
TIGR01855 |
imidazole glycerol phosphate synthase, glutamine amidotransferase subunit; This model ... |
66-186 |
4.27e-10 |
|
imidazole glycerol phosphate synthase, glutamine amidotransferase subunit; This model represents the glutamine amidotransferase subunit (or domain, in eukaryotic systems) of imidazole glycerol phosphate synthase. This subunit catalyzes step 5 of histidine biosynthesis from PRPP. The other subunit, the cyclase, catalyzes step 6. [Amino acid biosynthesis, Histidine family]
Pssm-ID: 273836 [Multi-domain] Cd Length: 196 Bit Score: 59.26 E-value: 4.27e-10
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 66 KISEDIFEIGVPVLGICYGHQLI---------CTTLG---GKV---ESAQVREYGKTDVVLNNSSGLFSGIDKNESCWMS 130
Cdd:TIGR01855 62 LFVELVVRLGKPVLGICLGMQLLferseegggVPGLGlikGNVvklEARKVPHMGWNEVHPVKESPLLNGIDEGAYFYFV 141
|
90 100 110 120 130 140
....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 131 HtdfvSY--PPEGFKIIGKS--GESPVAAVEniDKKIYGVQFHPEVEHTpFGKKMLSNFL 186
Cdd:TIGR01855 142 H----SYyaVCEEEAVLAYAdyGEKFPAAVQ--KGNIFGTQFHPEKSGK-TGLKLLENFL 194
|
|
| GAT_1 |
cd03128 |
Type 1 glutamine amidotransferase (GATase1)-like domain; Type 1 glutamine amidotransferase ... |
6-88 |
6.48e-10 |
|
Type 1 glutamine amidotransferase (GATase1)-like domain; Type 1 glutamine amidotransferase (GATase1)-like domain. This group contains proteins similar to Class I glutamine amidotransferases, the intracellular PH1704 from Pyrococcus horikoshii, the C-terminal of the large catalase: Escherichia coli HP-II, Sinorhizobium meliloti Rm1021 ThuA, the A4 beta-galactosidase middle domain and peptidase E. The majority of proteins in this group have a reactive Cys found in the sharp turn between a beta strand and an alpha helix termed the nucleophile elbow. For Class I glutamine amidotransferases proteins which transfer ammonia from the amide side chain of glutamine to an acceptor substrate, this Cys forms a Cys-His-Glu catalytic triad in the active site. Glutamine amidotransferases activity can be found in a range of biosynthetic enzymes included in this cd: glutamine amidotransferase, formylglycinamide ribonucleotide, GMP synthetase, anthranilate synthase component II, glutamine-dependent carbamoyl phosphate synthase (CPSase), cytidine triphosphate synthetase, gamma-glutamyl hydrolase, imidazole glycerol phosphate synthase and, cobyric acid synthase. For Pyrococcus horikoshii PH1704, the Cys of the nucleophile elbow together with a different His and, a Glu from an adjacent monomer form a catalytic triad different from the typical GATase1 triad. Peptidase E is believed to be a serine peptidase having a Ser-His-Glu catalytic triad which differs from the Cys-His-Glu catalytic triad of typical GATase1 domains, by having a Ser in place of the reactive Cys at the nucleophile elbow. The E. coli HP-II C-terminal domain, S. meliloti Rm1021 ThuA and the A4 beta-galactosidase middle domain lack the catalytic triad typical GATaseI domains. GATase1-like domains can occur either as single polypeptides, as in Class I glutamine amidotransferases, or as domains in a much larger multifunctional synthase protein, such as CPSase. Peptidase E has a circular permutation in the common core of a typical GTAse1 domain.
Pssm-ID: 153222 [Multi-domain] Cd Length: 92 Bit Score: 56.05 E-value: 6.48e-10
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 6 VIVVDFGGQYN---QLIARRVRENNVYCEIVPYT--YSVDKIKEKNPRGIIFTGGPNSVYD--DNAPKISE--DIFEIGV 76
Cdd:cd03128 1 VAVLLFGGSEElelASPLDALREAGAEVDVVSPDggPVESDVDLDDYDGLILPGGPGTPDDlaWDEALLALlrEAAAAGK 80
|
90
....*....|..
gi 219567478 77 PVLGICYGHQLI 88
Cdd:cd03128 81 PVLGICLGAQLL 92
|
|
| GATase1_IGP_Synthase |
cd01748 |
Type 1 glutamine amidotransferase (GATase1) domain found in imidazole glycerol phosphate ... |
75-186 |
7.32e-10 |
|
Type 1 glutamine amidotransferase (GATase1) domain found in imidazole glycerol phosphate synthase (IGPS); Type 1 glutamine amidotransferase (GATase1) domain found in imidazole glycerol phosphate synthase (IGPS). IGPS incorporates ammonia derived from glutamine into N1-[(5'-phosphoribulosyl)-formimino]-5-aminoimidazole-4-carboxamide ribonucleotide (PRFAR) to form 5'-(5-aminoimidazole-4-carboxamide) ribonucleotide (AICAR) and imidazole glycerol phosphate (IGP). The glutamine amidotransferase domain generates the ammonia nucleophile which is channeled from the glutaminase active site to the PRFAR active site. IGPS belong to the triad family of amidotransferases having a conserved Cys-His-Glu catalytic triad in the glutaminase active site.
Pssm-ID: 153219 [Multi-domain] Cd Length: 198 Bit Score: 58.66 E-value: 7.32e-10
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 75 GVPVLGICYGHQLICTT---------LG---GKVESAQVREYGKT------DVVLNNSSGLFSGIDKNescwmSHTDFV- 135
Cdd:cd01748 71 GKPFLGICLGMQLLFESseegggtkgLGlipGKVVRFPASEGLKVphmgwnQLEITKESPLFKGIPDG-----SYFYFVh 145
|
90 100 110 120 130
....*....|....*....|....*....|....*....|....*....|....*.
gi 219567478 136 SY---PPEGFKIIGKS--GESPVAAVENidKKIYGVQFHPEVEHTpFGKKMLSNFL 186
Cdd:cd01748 146 SYyapPDDPDYILATTdyGGKFPAAVEK--DNIFGTQFHPEKSGK-AGLKLLKNFL 198
|
|
| GATase1_2 |
cd01745 |
Subgroup of proteins having the Type 1 glutamine amidotransferase (GATase1) domain; This group ... |
32-171 |
7.71e-10 |
|
Subgroup of proteins having the Type 1 glutamine amidotransferase (GATase1) domain; This group contains a subgroup of proteins having the Type 1 glutamine amidotransferase (GATase1) domain. GATase activity catalyses the transfer of ammonia from the amide side chain of glutamine to an acceptor substrate. Glutamine amidotransferases (GATase) includes the triad family of amidotransferases which have a conserved Cys-His-Glu catalytic triad in the glutaminase active site. In this subgroup this triad is conserved. GATase activity can be found in a range of biosynthetic enzymes, including: glutamine amidotransferase, formylglycinamide ribonucleotide, GMP synthetase , anthranilate synthase component II, glutamine-dependent carbamoyl phosphate synthase, cytidine triphosphate synthetase, gamma-glutamyl hydrolase, imidazole glycerol phosphate synthase and, cobyric acid synthase. Glutamine amidotransferase (GATase) domains can occur either as single polypeptides, as in glutamine amidotransferases, or as domains in a much larger multifunctional synthase protein, such as CPSase.
Pssm-ID: 153216 [Multi-domain] Cd Length: 189 Bit Score: 58.36 E-value: 7.71e-10
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 32 IVPYTYSVDKIKE--KNPRGIIFTGGPN----SVYDDNAPKISE-----DIFEI---------GVPVLGICYGHQLICTT 91
Cdd:cd01745 37 LLPPVDDEEDLEQylELLDGLLLTGGGDvdppLYGEEPHPELGPidperDAFELallraalerGKPILGICRGMQLLNVA 116
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 92 LGGKVEsaQvreygktDVVLNnsSGLFSGIDKnescwmshtdfvsyPPEGFKIIGKSGESPVAAVENIDKK-IYGVQFHP 170
Cdd:cd01745 117 LGGTLY--Q-------DIRVN--SLHHQAIKR--------------LADGLRVEARAPDGVIEAIESPDRPfVLGVQWHP 171
|
.
gi 219567478 171 E 171
Cdd:cd01745 172 E 172
|
|
| PRK06774 |
PRK06774 |
aminodeoxychorismate synthase component II; |
42-186 |
9.87e-10 |
|
aminodeoxychorismate synthase component II;
Pssm-ID: 180689 [Multi-domain] Cd Length: 191 Bit Score: 57.95 E-value: 9.87e-10
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 42 IKEKNPRGIIFTGGPNSvydDNAPKISEDI---FEIGVPVLGICYGHQLICTTLGGKVESAQVREYGKTDVVLNNSSGLF 118
Cdd:PRK06774 39 IEQLAPSHLVISPGPCT---PNEAGISLAVirhFADKLPILGVCLGHQALGQAFGARVVRARQVMHGKTSAICHSGQGVF 115
|
90 100 110 120 130 140 150
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....
gi 219567478 119 SGIDKNESCWMSHTDFVSYP--PEGFKIIGKSGES----PVAAVENIDKKIYGVQFHPEVEHTPFGKKMLSNFL 186
Cdd:PRK06774 116 RGLNQPLTVTRYHSLVIAADslPGCFELTAWSERGgemdEIMGIRHRTLPLEGVQFHPESILSEQGHQLLDNFL 189
|
|
| LarE-like |
cd01990 |
Lactate racemization operon protein LarE and similar proteins; This subfamily includes ... |
217-407 |
9.94e-10 |
|
Lactate racemization operon protein LarE and similar proteins; This subfamily includes Lactiplantibacillus plantarum LarE, a sacrificial sulfur insertase of the N-type ATP pyrophosphatase family. LarE is part of the lar operon, encoding five Lar proteins (LarA-E) that collaboratively synthesize and incorporate a niacin-derived Ni-containing cofactor into LarA, an Ni-dependent lactate racemase. It catalyzes successive thiolation reactions by donating the sulfur atom of their exclusive cysteine residues to the substrate. The LarE-like subfamily belongs to the nucleotide alpha hydrolase (AANH) superfamily that includes N-type ATP PPases and ATP sulfurylases. It forms an alpha/beta/alpha fold which binds to the adenosine group. Proteins from this subfamily probably binds ATP. This domain is about 200 amino acids long with a strongly conserved motif SGGxDS at the N-terminus.
Pssm-ID: 467494 [Multi-domain] Cd Length: 222 Bit Score: 58.81 E-value: 9.94e-10
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 217 KVICAMSGGVDSSVAAMIVHKAVGKQLTCIFVDHGLLRKDEGDQVEDIFKKqfnmnfIRVNAEKRFLQKLKD---ISDPE 293
Cdd:cd01990 1 KVVVAFSGGVDSSLLAKLAKEVLGDNVVAVTADSPLVPREELEEAKRIAEE------IGIRHEIIKTDELDDeeyVANDP 74
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 294 KK----RKIIGEEFIrvfeEEAKKLGeIAFLVQGTIYPDVVESGLGTSATIKSHHNVgglpedmdfkliePLRE--LFKD 367
Cdd:cd01990 75 DRcyhcKKALYSTLK----EIAKERG-YDVVLDGTNADDLKDYRPGLLAAAELGIRS-------------PLPElgLTKS 136
|
170 180 190 200
....*....|....*....|....*....|....*....|..
gi 219567478 368 EVRAVGEELGIPhklVWRQPfPGPGLAIRVLGN--ITEEKLQ 407
Cdd:cd01990 137 EIRELARELGLP---NWDKP-ASACLASRIPYGeeITPERLK 174
|
|
| hisH |
PRK13141 |
imidazole glycerol phosphate synthase subunit HisH; Provisional |
75-186 |
1.13e-09 |
|
imidazole glycerol phosphate synthase subunit HisH; Provisional
Pssm-ID: 237288 [Multi-domain] Cd Length: 205 Bit Score: 58.22 E-value: 1.13e-09
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 75 GVPVLGICYGHQLI---------CTTLG---GKVESAQVREYGK------TDVVLNNSSGLFSGIDKNescwmSHTDFV- 135
Cdd:PRK13141 72 GKPLLGICLGMQLLfesseefgeTEGLGllpGRVRRFPPEEGLKvphmgwNQLELKKESPLLKGIPDG-----AYVYFVh 146
|
90 100 110 120 130
....*....|....*....|....*....|....*....|....*....|....*.
gi 219567478 136 SY---PPEGFKIIGKS--GESPVAAVENidKKIYGVQFHPEVEHTPfGKKMLSNFL 186
Cdd:PRK13141 147 SYyadPCDEEYVAATTdyGVEFPAAVGK--DNVFGAQFHPEKSGDV-GLKILKNFV 199
|
|
| PRK09522 |
PRK09522 |
bifunctional anthranilate synthase glutamate amidotransferase component TrpG/anthranilate ... |
15-186 |
2.09e-09 |
|
bifunctional anthranilate synthase glutamate amidotransferase component TrpG/anthranilate phosphoribosyltransferase TrpD;
Pssm-ID: 181927 [Multi-domain] Cd Length: 531 Bit Score: 59.65 E-value: 2.09e-09
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 15 YNqlIARRVRENN----VYCEIVPYTYSVDKIKEKNPRGIIFTGGPNSVYDdnAPKISEDIFEI--GVPVLGICYGHQLI 88
Cdd:PRK09522 15 YN--LADQLRSNGhnvvIYRNHIPAQTLIERLATMSNPVLMLSPGPGVPSE--AGCMPELLTRLrgKLPIIGICLGHQAI 90
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 89 CTTLGGKVESAQVREYGKTDVVLNNSSGLFSGIDKNESCWMSHTDFVSYPPEGFKiIGKSGESPVAAVENIDKKIYGVQF 168
Cdd:PRK09522 91 VEAYGGYVGQAGEILHGKASSIEHDGQAMFAGLTNPLPVARYHSLVGSNIPAGLT-INAHFNGMVMAVRHDADRVCGFQF 169
|
170
....*....|....*...
gi 219567478 169 HPEVEHTPFGKKMLSNFL 186
Cdd:PRK09522 170 HPESILTTQGARLLEQTL 187
|
|
| hisH |
PRK13143 |
imidazole glycerol phosphate synthase subunit HisH; Provisional |
49-186 |
1.99e-08 |
|
imidazole glycerol phosphate synthase subunit HisH; Provisional
Pssm-ID: 237289 [Multi-domain] Cd Length: 200 Bit Score: 54.49 E-value: 1.99e-08
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 49 GIIFTG-GPNSVYDDNAPKISEDIFEI---GVPVLGICYGHQLICTT---------LG---GKV----ESAQVREYGKTD 108
Cdd:PRK13143 41 GIVLPGvGAFGAAMENLSPLRDVILEAarsGKPFLGICLGMQLLFESseegggvrgLGlfpGRVvrfpAGVKVPHMGWNT 120
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 109 VVLNNSSGLFSGIDkNESCWMSHTdFVSYPPEGFKIIGKS--GESPVAAVENidKKIYGVQFHPEVEHTPfGKKMLSNFL 186
Cdd:PRK13143 121 VKVVKDCPLFEGID-GEYVYFVHS-YYAYPDDEDYVVATTdyGIEFPAAVCN--DNVFGTQFHPEKSGET-GLKILENFV 195
|
|
| hisH |
PRK13146 |
imidazole glycerol phosphate synthase subunit HisH; Provisional |
75-186 |
3.29e-08 |
|
imidazole glycerol phosphate synthase subunit HisH; Provisional
Pssm-ID: 237290 [Multi-domain] Cd Length: 209 Bit Score: 54.02 E-value: 3.29e-08
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 75 GVPVLGICYGHQLICTT------------LGGKVESAQVRE-------YGKTDVVLNNSSGLFSGIDKNEscwmsHTDFV 135
Cdd:PRK13146 77 GRPFLGICVGMQLLFERglehgdtpglglIPGEVVRFQPDGpalkvphMGWNTVDQTRDHPLFAGIPDGA-----RFYFV 151
|
90 100 110 120 130
....*....|....*....|....*....|....*....|....*....|....*..
gi 219567478 136 -SY---PPEGFKIIGKS--GESPVAAVENidKKIYGVQFHPEVEHTpFGKKMLSNFL 186
Cdd:PRK13146 152 hSYyaqPANPADVVAWTdyGGPFTAAVAR--DNLFATQFHPEKSQD-AGLALLRNFL 205
|
|
| hisH |
PRK13170 |
imidazole glycerol phosphate synthase subunit HisH; Provisional |
77-186 |
3.79e-08 |
|
imidazole glycerol phosphate synthase subunit HisH; Provisional
Pssm-ID: 183877 [Multi-domain] Cd Length: 196 Bit Score: 53.32 E-value: 3.79e-08
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 77 PVLGICYGHQLI---------CTTLG---GKVESAQVREY-----GKTDVVLNNSSGLFSGIDKNescwmSHTDFV-SY- 137
Cdd:PRK13170 72 PVLGICLGMQLLgerseesggVDCLGiidGPVKKMTDFGLplphmGWNQVTPQAGHPLFQGIEDG-----SYFYFVhSYa 146
|
90 100 110 120 130
....*....|....*....|....*....|....*....|....*....|.
gi 219567478 138 -PPEGFKI-IGKSGESPVAAVENidKKIYGVQFHPEVEHTPfGKKMLSNFL 186
Cdd:PRK13170 147 mPVNEYTIaQCNYGEPFSAAIQK--DNFFGVQFHPERSGAA-GAQLLKNFL 194
|
|
| HisH |
COG0118 |
Imidazoleglycerol phosphate synthase glutamine amidotransferase subunit HisH [Amino acid ... |
69-171 |
1.08e-07 |
|
Imidazoleglycerol phosphate synthase glutamine amidotransferase subunit HisH [Amino acid transport and metabolism]; Imidazoleglycerol phosphate synthase glutamine amidotransferase subunit HisH is part of the Pathway/BioSystem: Histidine biosynthesis
Pssm-ID: 439888 [Multi-domain] Cd Length: 196 Bit Score: 51.96 E-value: 1.08e-07
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 69 EDIFEIGVPVLGICYGHQLICT---------TLG---GKVESAQVREY-----GKTDVVLNNSSGLFSGIDKNEscwmsH 131
Cdd:COG0118 67 REAVAGGKPVLGICLGMQLLFErseengdteGLGlipGEVVRFPASDLkvphmGWNTVEIAKDHPLFAGIPDGE-----Y 141
|
90 100 110 120
....*....|....*....|....*....|....*....|....*.
gi 219567478 132 TDFV-SY--PPEGFK-IIGKS--GESPVAAVENidKKIYGVQFHPE 171
Cdd:COG0118 142 FYFVhSYyvPPDDPEdVVATTdyGVPFTAAVER--GNVFGTQFHPE 185
|
|
| tRNA_Me_trans |
pfam03054 |
tRNA methyl transferase HUP domain; This family represents the N-terminal HUP domain in tRNA ... |
216-379 |
1.33e-07 |
|
tRNA methyl transferase HUP domain; This family represents the N-terminal HUP domain in tRNA(5-methylaminomethyl-2-thiouridine)-methyltransferase which is involved in the biosynthesis of the modified nucleoside 5-methylaminomethyl-2-thiouridine present in the wobble position of some tRNAs.
Pssm-ID: 460787 [Multi-domain] Cd Length: 202 Bit Score: 51.87 E-value: 1.33e-07
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 216 KKVICAMSGGVDSSVAAMIVHKAvGKQLTCIFV---DHGLLRKDEG-----DQVEDIFK--KQFNMNFIRVNAEKRFlqk 285
Cdd:pfam03054 1 MKVVVAMSGGVDSSVAAYLLKEQ-GHNVIGVFMknwDEEQSLDEEGkccseEDLADAQRvcEQLGIPLYVVNFEKEY--- 76
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 286 lkdisdpekKRKIIgEEFIRVFEE-----------EAKKLGeiAFLvqgtiypDVVESGLGTSATIKSH-----HNVGGL 349
Cdd:pfam03054 77 ---------WEDVF-EPFLDEYKNgrtpnpdvlcnKEIKFG--ALL-------DYALENLGADYVATGHyarvsLNKDGG 137
|
170 180 190 200 210
....*....|....*....|....*....|....*....|....*....|
gi 219567478 350 PE-----DMD-----F----------KLIEPLRELFKDEVRAVGEELGIP 379
Cdd:pfam03054 138 SEllralDKNkdqsyFlstlsqeqleKLLFPLGELTKEEVRKIAKEAGLA 187
|
|
| PRK13566 |
PRK13566 |
anthranilate synthase component I; |
75-171 |
1.46e-07 |
|
anthranilate synthase component I;
Pssm-ID: 237429 [Multi-domain] Cd Length: 720 Bit Score: 54.15 E-value: 1.46e-07
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 75 GVPVLGICYGHQLICTTLGGKVESAQVREYGKTDVVLNN-SSGLFSGIDKNESCWMSHTDFVSYP--PEGFKIIGKSGES 151
Cdd:PRK13566 598 NLPIFGVCLGLQAIVEAFGGELGQLAYPMHGKPSRIRVRgPGRLFSGLPEEFTVGRYHSLFADPEtlPDELLVTAETEDG 677
|
90 100
....*....|....*....|
gi 219567478 152 PVAAVENIDKKIYGVQFHPE 171
Cdd:PRK13566 678 VIMAIEHKTLPVAAVQFHPE 697
|
|
| PuuD |
COG2071 |
Gamma-glutamyl-gamma-aminobutyrate hydrolase PuuD (putrescine degradation), contains ... |
49-185 |
3.65e-07 |
|
Gamma-glutamyl-gamma-aminobutyrate hydrolase PuuD (putrescine degradation), contains GATase1-like domain [Amino acid transport and metabolism];
Pssm-ID: 441674 [Multi-domain] Cd Length: 231 Bit Score: 51.32 E-value: 3.65e-07
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 49 GIIFTGGPN---SVYD-DNAPKISE-----DIFEI---------GVPVLGICYGHQLICTTLGG--------KVESA--- 99
Cdd:COG2071 52 GLVLTGGADvdpALYGeEPHPELGPidperDAFELaliraalerGKPVLGICRGMQLLNVALGGtlyqdlpdQVPGAldh 131
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 100 ---QVREYGKTDVVLNNSSGLFS---------------GIDKnescwmshtdfvsyPPEGFKIIGKSG----EspvaAVE 157
Cdd:COG2071 132 rqpAPRYAPRHTVEIEPGSRLARilgeeeirvnslhhqAVKR--------------LGPGLRVSARAPdgviE----AIE 193
|
170 180 190
....*....|....*....|....*....|.
gi 219567478 158 NIDKK-IYGVQFHPE--VEHTPFGKKMLSNF 185
Cdd:COG2071 194 SPGAPfVLGVQWHPEwlAASDPLSRRLFEAF 224
|
|
| mnmA |
PRK00143 |
tRNA-specific 2-thiouridylase MnmA; Reviewed |
216-305 |
4.93e-07 |
|
tRNA-specific 2-thiouridylase MnmA; Reviewed
Pssm-ID: 234664 [Multi-domain] Cd Length: 346 Bit Score: 51.61 E-value: 4.93e-07
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 216 KKVICAMSGGVDSSVAAMIVHKAvGKQLTCIFV---DHGLLRKDEGD-QVEDIFK-----KQFNMNFIRVNAEKRFlqkl 286
Cdd:PRK00143 1 KRVVVGMSGGVDSSVAAALLKEQ-GYEVIGVFMklwDDDDETGKGGCcAEEDIADarrvaDKLGIPHYVVDFEKEF---- 75
|
90
....*....|....*....
gi 219567478 287 kdisdpekKRKIIgEEFIR 305
Cdd:PRK00143 76 --------WDRVI-DYFLD 85
|
|
| MnmA |
COG0482 |
tRNA U34 2-thiouridine synthase MnmA/TrmU, contains the PP-loop ATPase domain [Translation, ... |
216-282 |
7.82e-07 |
|
tRNA U34 2-thiouridine synthase MnmA/TrmU, contains the PP-loop ATPase domain [Translation, ribosomal structure and biogenesis]; tRNA U34 2-thiouridine synthase MnmA/TrmU, contains the PP-loop ATPase domain is part of the Pathway/BioSystem: tRNA modification
Pssm-ID: 440250 [Multi-domain] Cd Length: 353 Bit Score: 51.21 E-value: 7.82e-07
10 20 30 40 50 60 70
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*.
gi 219567478 216 KKVICAMSGGVDSSVAAMIVHKAvGKQLTCIFV---DHGLLRKDEGD-QVEDIFK-----KQFNMNFIRVNAEKRF 282
Cdd:COG0482 1 KRVVVGMSGGVDSSVAAALLKEQ-GYEVIGVTMklwDDDDASGSGGCcSLEDIEDarrvaDKLGIPHYVVDFEEEF 75
|
|
| COG1606 |
COG1606 |
ATP-utilizing enzyme, PP-loop superfamily [General function prediction only]; |
202-407 |
2.36e-06 |
|
ATP-utilizing enzyme, PP-loop superfamily [General function prediction only];
Pssm-ID: 441214 [Multi-domain] Cd Length: 265 Bit Score: 48.95 E-value: 2.36e-06
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 202 VDEKIKSIKEEVGD-KKVICAMSGGVDSSVAAMIVHKAVGKQLTCIFVDHGLLRKDEGDQVEDiFKKQFNMNFIRVNAEk 280
Cdd:COG1606 1 LEEKLERLKAILKElGSVLVAFSGGVDSTLLAKVAHDVLGDRVLAVTADSPSLPERELEEAKE-LAKEIGIRHEVIETD- 78
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 281 rflqklkDISDPE------------KKrkiigeEFIRVFEEEAKKLGeIAFLVQGTIYPD----------VVESGlgtsa 338
Cdd:COG1606 79 -------ELEDPEfvanppdrcyhcKK------ELFSKLKELAKELG-YAVVADGTNADDlgdyrpglraAKELG----- 139
|
170 180 190 200 210 220 230
....*....|....*....|....*....|....*....|....*....|....*....|....*....|...
gi 219567478 339 tIKShhnvgglpedmdfkliePLRE--LFKDEVRAVGEELGIPhklVWRQPfPGPGLAIRVLGN--ITEEKLQ 407
Cdd:COG1606 140 -VRS-----------------PLAEagLTKAEIRELARELGLP---TWDKP-SSACLASRIPYGeeITPEKLR 190
|
|
| hisH |
PRK13152 |
imidazole glycerol phosphate synthase subunit HisH; Provisional |
67-186 |
3.12e-06 |
|
imidazole glycerol phosphate synthase subunit HisH; Provisional
Pssm-ID: 171876 [Multi-domain] Cd Length: 201 Bit Score: 47.91 E-value: 3.12e-06
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 67 ISEDIFEIGVPVLGICYGHQLICT--TLGGKVES-----AQVREY-----------GKTDVVLNNSSGLFSGIDKNESCW 128
Cdd:PRK13152 65 LKEQVLVQKKPILGICLGMQLFLErgYEGGVCEGlgfieGEVVKFeedlnlkiphmGWNELEILKQSPLYQGIPEKSDFY 144
|
90 100 110 120 130
....*....|....*....|....*....|....*....|....*....|....*...
gi 219567478 129 MSHTDFVSYPPEGFKIIGKSGESPVAAVENidKKIYGVQFHPEVEHTpFGKKMLSNFL 186
Cdd:PRK13152 145 FVHSFYVKCKDEFVSAKAQYGHKFVASLQK--DNIFATQFHPEKSQN-LGLKLLENFA 199
|
|
| PRK07053 |
PRK07053 |
glutamine amidotransferase; Provisional |
54-105 |
5.09e-06 |
|
glutamine amidotransferase; Provisional
Pssm-ID: 235919 [Multi-domain] Cd Length: 234 Bit Score: 47.63 E-value: 5.09e-06
10 20 30 40 50
....*....|....*....|....*....|....*....|....*....|....*....
gi 219567478 54 GGPNSVYDDNA-PKISEDI------FEIGVPVLGICYGHQLICTTLGGKVESAQVREYG 105
Cdd:PRK07053 55 GGPIGVYDDELyPFLAPEIallrqrLAAGLPTLGICLGAQLIARALGARVYPGGQKEIG 113
|
|
| NAD_synthase |
cd00553 |
NAD(+) synthase; NAD(+) synthase (EC 6.3.5.1), a homodimer, catalyzes the final step in the de ... |
214-409 |
6.22e-06 |
|
NAD(+) synthase; NAD(+) synthase (EC 6.3.5.1), a homodimer, catalyzes the final step in the de novo nicotinamide adenine dinucleotide (NAD(+)) biosynthesis, an amide transfer from either ammonia or glutamine to nicotinic acid adenine dinucleotide (NaAD). The conversion of NaAD to NAD(+) occurs via a NAD-adenylate intermediate and requires ATP and Mg(2+). The intermediate is subsequently cleaved into NAD(+) and AMP. In many prokaryotes, such as Escherichia coli, NAD synthase consists of a single domain and is strictly ammonia dependent. In contrast, eukaryotes and other prokaryotes have an additional N-terminal amidohydrolase domain that prefer glutamine. Interestingly, NAD(+) synthases in these prokaryotes, can also utilize ammonia as an amide source.
Pssm-ID: 467484 [Multi-domain] Cd Length: 248 Bit Score: 47.55 E-value: 6.22e-06
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 214 GDKKVICAMSGGVDSSVAAMIVHKAVGK-QLTCIFVDHGLLRKDEGDQVEDiFKKQFNMNFIRVN---AEKRFLQKLKDI 289
Cdd:cd00553 22 GAKGFVLGLSGGIDSAVVAALAVRALGAeNVLALIMPSRYSSKETRDDAKA-LAENLGIEYRTIDidpIVDAFLKALEHA 100
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 290 SDPEKKRKIIG-----EEFIRVFeEEAKKLGeiaFLVQGTiyPDVVESGLGTsATIkshhnvGGlpeDM--DfklIEPLR 362
Cdd:cd00553 101 GGSEAEDLALGniqarLRMVLLY-ALANLLG---GLVLGT--GNKSELLLGY-FTK------YG---DGaaD---INPIG 161
|
170 180 190 200
....*....|....*....|....*....|....*....|....*..
gi 219567478 363 ELFKDEVRAVGEELGIPHKlVWRQPfPGPGLAIrvlGNITEEKLQIT 409
Cdd:cd00553 162 DLYKTQVRELARYLGVPEE-IIEKP-PSAELWP---GQTDEDELGMP 203
|
|
| PRK06490 |
PRK06490 |
glutamine amidotransferase; Provisional |
50-174 |
8.02e-06 |
|
glutamine amidotransferase; Provisional
Pssm-ID: 180590 [Multi-domain] Cd Length: 239 Bit Score: 47.26 E-value: 8.02e-06
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 50 IIFtGGPNSVYDDNaPKISEDIFEIGVPV------LGICYGHQLICTTLGGKVESaqvREYGKTDV------VLNNSSGL 117
Cdd:PRK06490 57 VIF-GGPMSANDPD-DFIRREIDWISVPLkenkpfLGICLGAQMLARHLGARVAP---HPDGRVEIgyyplrPTEAGRAL 131
|
90 100 110 120 130 140
....*....|....*....|....*....|....*....|....*....|....*....|....*
gi 219567478 118 FSgidknescWMSHTdfvsYP--------PEGFKIIGKSGESPVAAVENiDKKIYGVQFHPEVEH 174
Cdd:PRK06490 132 MH--------WPEMV----YHwhregfdlPAGAELLATGDDFPNQAFRY-GDNAWGLQFHPEVTR 183
|
|
| PRK05637 |
PRK05637 |
anthranilate synthase component II; Provisional |
30-183 |
8.70e-06 |
|
anthranilate synthase component II; Provisional
Pssm-ID: 180178 [Multi-domain] Cd Length: 208 Bit Score: 46.76 E-value: 8.70e-06
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 30 CEIVPYTYSVDKIKEKNPRGIIFTGGPNsvYDDNAPKISEDIFE-IG-VPVLGICYGHQLICTTLGGKVESAQvREYGKT 107
Cdd:PRK05637 28 CTVFRNTVPVEEILAANPDLICLSPGPG--HPRDAGNMMALIDRtLGqIPLLGICLGFQALLEHHGGKVEPCG-PVHGTT 104
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 108 D-VVLNNS---SGLFSGID-----KNESCWMSHTDFVSY-------PPEGFKIIG--KSGESPVA-AVENIDKKIYGVQF 168
Cdd:PRK05637 105 DnMILTDAgvqSPVFAGLAtdvepDHPEIPGRKVPIARYhslgcvvAPDGMESLGtcSSEIGPVImAAETTDGKAIGLQF 184
|
170
....*....|....*
gi 219567478 169 HPEVEHTPFGKKMLS 183
Cdd:PRK05637 185 HPESVLSPTGPIILS 199
|
|
| PRK07567 |
PRK07567 |
glutamine amidotransferase; Provisional |
49-171 |
1.33e-05 |
|
glutamine amidotransferase; Provisional
Pssm-ID: 181035 [Multi-domain] Cd Length: 242 Bit Score: 46.47 E-value: 1.33e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 49 GIIFTGGPNSVYDDNAPK-------------ISEDIFEIGVPVLGICYGHQLICTTLGGKVESAQVREYGKTDVVLNNSS 115
Cdd:PRK07567 54 GVIVGGSPFNVSDPAESKspwqrrveaelsgLLDEVVARDFPFLGACYGVGTLGHHQGGVVDRTYGEPVGAVTVSLTDAG 133
|
90 100 110 120 130
....*....|....*....|....*....|....*....|....*....|....*....
gi 219567478 116 G---LFSGIDKNESCWMSHTDFVSYPPEGFKIIGKSGESPVAAVEnIDKKIYGVQFHPE 171
Cdd:PRK07567 134 RadpLLAGLPDTFTAFVGHKEAVSALPPGAVLLATSPTCPVQMFR-VGENVYATQFHPE 191
|
|
| MnmA_TRMU-like |
cd01998 |
MnmA/TRMU family 2-thiouridylases and similar proteins; This family is composed of bacterial ... |
217-282 |
1.49e-05 |
|
MnmA/TRMU family 2-thiouridylases and similar proteins; This family is composed of bacterial tRNA-specific 2-thiouridylase MnmA (EC 2.8.1.13) and mitochondrial tRNA-specific 2-thiouridylase 1 (TRMU or MTU1, EC 2.8.1.14). MnmA catalyzes the 2-thiolation of uridine at the wobble position (U34) of tRNA, leading to the formation of s(2)U34. TRMU/MTU1 catalyzes the 2-thiolation of uridine at the wobble position (U34) of mitochondrial tRNA(Lys), tRNA(Glu) and tRNA(Gln); this is required for the formation of 5-taurinomethyl-2-thiouridine (tm5s2U) of mitochondrial tRNA(Lys), tRNA(Glu), and tRNA(Gln) at the wobble position. This family belongs to the adenine nucleotide alpha hydrolase (AANH) superfamily that also includes other N-type ATP PPases and ATP sulfurylases. It forms an alpha/beta/alpha fold which binds to the adenosine group.
Pssm-ID: 467502 [Multi-domain] Cd Length: 349 Bit Score: 47.12 E-value: 1.49e-05
10 20 30 40 50 60 70
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....
gi 219567478 217 KVICAMSGGVDSSVAAMIVHKAvGKQLTCIFV---DHGLLRKDE---GDQVEDIFK--KQFNMNFIRVNAEKRF 282
Cdd:cd01998 1 KVAVAMSGGVDSSVAAALLKEQ-GYDVIGVFMknwDDEDNEKGGccsEEDIEDARRvaDQLGIPLYVVDFSEEY 73
|
|
| NAD_synthase |
pfam02540 |
NAD synthase; NAD synthase (EC:6.3.5.1) is involved in the de novo synthesis of NAD and is ... |
206-432 |
8.84e-05 |
|
NAD synthase; NAD synthase (EC:6.3.5.1) is involved in the de novo synthesis of NAD and is induced by stress factors such as heat shock and glucose limitation.
Pssm-ID: 396888 [Multi-domain] Cd Length: 241 Bit Score: 43.91 E-value: 8.84e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 206 IKSIKEEVGDKKVICAMSGGVDSSVAAMIVHKAVGKQLTcifvdHGLL----RKDEGDqVEDI--FKKQFNMNFIRVNAE 279
Cdd:pfam02540 9 LRDYVQKAGFKGVVLGLSGGIDSSLVAYLAVKALGKENV-----LALImpssQSSEED-VQDAlaLAENLGIEYKTIDIK 82
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 280 ---KRFLQKLKDISDPEKKRKIIGEEFIRVFEEEAKKLGeiaFLVQGTIYPDVVESGLGTSatikshhnVGGLPEDmdfk 356
Cdd:pfam02540 83 pivRAFSQLFQDASEDFAKGNLKARIRMAILYYIANKFN---YLVLGTGNKSELAVGYFTK--------YGDGACD---- 147
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 357 lIEPLRELFKDEVRAVGEELGIPHKLVWRQP----FPgpglairvlGNITEEKLQITRDA-DAIF--------REEIAKA 423
Cdd:pfam02540 148 -IAPIGDLYKTQVYELARYLNVPERIIKKPPsadlWP---------GQTDEEELGIPYDElDDILklvekklsPEEIIGK 217
|
....*....
gi 219567478 424 NLDETIWQY 432
Cdd:pfam02540 218 GLPAEVVRR 226
|
|
| AANH-like |
cd01986 |
adenine nucleotide alpha hydrolase (AANH)-like proteins; This group of adenine nucleotide ... |
218-283 |
1.02e-04 |
|
adenine nucleotide alpha hydrolase (AANH)-like proteins; This group of adenine nucleotide alpha hydrolase (AANH)-like proteins includes N-type ATP PPases and ATP sulfurylases. The domain forms an alpha/beta/alpha fold which binds to adenosine nucleotide.
Pssm-ID: 467490 [Multi-domain] Cd Length: 74 Bit Score: 40.51 E-value: 1.02e-04
10 20 30 40 50 60
....*....|....*....|....*....|....*....|....*....|....*....|....*..
gi 219567478 218 VICAMSGGVDSSVAAMIVHKAVGK-QLTCIFVDHGLLRKDEGDQVEDIFKKQFnMNFIRVNAEKRFL 283
Cdd:cd01986 1 VVVGYSGGKDSSVALHLASRLGRKaEVAVVHIDHGIGFKEEAESVASIARRSI-LKKLAEKGARAIA 66
|
|
| TilS_N |
cd01992 |
N-terminal domain of tRNA(Ile)-lysidine synthase and similar proteins; tRNA(Ile)-lysidine ... |
217-379 |
1.16e-04 |
|
N-terminal domain of tRNA(Ile)-lysidine synthase and similar proteins; tRNA(Ile)-lysidine synthase (EC 6.3.4.19), also called tRNA(Ile)-2-lysyl-cytidine synthase or tRNA(Ile)-lysidine synthetase, catalyzes the ligation of lysine onto the cytidine present at position 34 of the AUA codon-specific tRNA(Ile) that contains the anticodon CAU, in an ATP-dependent manner. Cytidine is converted to lysidine, thus changing the amino acid specificity of the tRNA from methionine to isoleucine. This subfamily belongs to the adenine nucleotide alpha hydrolase superfamily that also includes N-type ATP PPases and ATP sulfurylases. It forms an alpha/beta/alpha fold which binds to adenosine group. This domain has a strongly conserved motif SGGXD at the N-terminus.
Pssm-ID: 467496 [Multi-domain] Cd Length: 185 Bit Score: 42.97 E-value: 1.16e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 217 KVICAMSGGVDSSV---AAMIVHKAVGKQLTCIFVDHGlLR---KDEGDQVEDIFkKQFNMNFIRVNAEKRFlqklKDIS 290
Cdd:cd01992 1 KILVAVSGGPDSMAllhLLKELRPKLGLKLVAVHVDHG-LReesAEEAQFVAKLC-KKLGIPLHILTVTEAP----KSGG 74
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 291 DPEKK-RKiigeefIR--VFEEEAKKLGeIAFLV----QGtiypDVVE---------SGLGTSATIKSHHNVGGLPedmd 354
Cdd:cd01992 75 NLEAAaRE------ARyaFLERAAKEHG-IDVLLtahhLD----DQAEtvlmrllrgSGLSGLAGMAARSKAGGIR---- 139
|
170 180
....*....|....*....|....*
gi 219567478 355 fkLIEPLRELFKDEVRAVGEELGIP 379
Cdd:cd01992 140 --LIRPLLGISKAELLAYCRENGLP 162
|
|
| trmU |
TIGR00420 |
tRNA (5-methylaminomethyl-2-thiouridylate)-methyltransferase; tRNA ... |
216-247 |
1.76e-04 |
|
tRNA (5-methylaminomethyl-2-thiouridylate)-methyltransferase; tRNA (5-methylaminomethyl-2-thiouridylate)-methyltransferase (trmU, asuE, or mnmA) is involved in the biosynthesis of the modified nucleoside 5-methylaminomethyl-2-thiouridine (mnm5s2U34) present in the wobble position of some tRNAs. This enzyme appears not to occur in the Archaea. [Protein synthesis, tRNA and rRNA base modification]
Pssm-ID: 273069 [Multi-domain] Cd Length: 352 Bit Score: 43.91 E-value: 1.76e-04
10 20 30
....*....|....*....|....*....|..
gi 219567478 216 KKVICAMSGGVDSSVAAMIVHKAvGKQLTCIF 247
Cdd:TIGR00420 1 KKVIVGLSGGVDSSVSAYLLKQQ-GYEVVGVF 31
|
|
| PRK13794 |
PRK13794 |
hypothetical protein; Provisional |
201-287 |
1.22e-03 |
|
hypothetical protein; Provisional
Pssm-ID: 237509 [Multi-domain] Cd Length: 479 Bit Score: 41.19 E-value: 1.22e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 201 FVDEKIKSIKeevgdKKVICAMSGGVDSSVAAMIVHKAVGKQLTCIFVDHGLLRKDEGDQVEDIfKKQFNMNFIRVNAEK 280
Cdd:PRK13794 238 FIRNTAEKIN-----KPVTVAYSGGKDSLATLLLALKALGINFPVLFNDTGLEFPETLENVEDV-EKHYGLEIIRTKSEE 311
|
....*..
gi 219567478 281 rFLQKLK 287
Cdd:PRK13794 312 -FWEKLE 317
|
|
| NadE |
COG0171 |
NH3-dependent NAD+ synthetase [Coenzyme transport and metabolism]; NH3-dependent NAD+ ... |
209-242 |
1.26e-03 |
|
NH3-dependent NAD+ synthetase [Coenzyme transport and metabolism]; NH3-dependent NAD+ synthetase is part of the Pathway/BioSystem: NAD biosynthesis
Pssm-ID: 439941 [Multi-domain] Cd Length: 542 Bit Score: 41.37 E-value: 1.26e-03
10 20 30
....*....|....*....|....*....|....*..
gi 219567478 209 IKEEV---GDKKVICAMSGGVDSSVAAMIVHKAVGKQ 242
Cdd:COG0171 277 LRDYVrknGFKGVVLGLSGGIDSALVAALAVDALGPE 313
|
|
| hisH |
PRK14004 |
imidazole glycerol phosphate synthase subunit HisH; Provisional |
35-186 |
1.40e-03 |
|
imidazole glycerol phosphate synthase subunit HisH; Provisional
Pssm-ID: 172505 [Multi-domain] Cd Length: 210 Bit Score: 40.27 E-value: 1.40e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 35 YTYSVDKIKEKNPRGIIFTGGPN---SVYDDNAPKISEDI---FEIGVPVLGICYGHQLIC-----TTLGGKVES----- 98
Cdd:PRK14004 26 FVFTSDPETIENSKALILPGDGHfdkAMENLNSTGLRSTIdkhVESGKPLFGICIGFQILFesseeTNQGTKKEQieglg 105
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 99 ---AQVREYGKTD------------VVLNNSSGLFSGIDKNESCWMSHtdfvSYPP---EGFKIIGKS---GESPVAAVE 157
Cdd:PRK14004 106 yikGKIKKFEGKDfkvphigwnrlqIRRKDKSKLLKGIGDQSFFYFIH----SYRPtgaEGNAITGLCdyyQEKFPAVVE 181
|
170 180
....*....|....*....|....*....
gi 219567478 158 NidKKIYGVQFHPEVEHTpFGKKMLSNFL 186
Cdd:PRK14004 182 K--ENIFGTQFHPEKSHT-HGLKLLENFI 207
|
|
| TilS |
COG0037 |
tRNA(Ile)-lysidine synthase TilS/MesJ [Translation, ribosomal structure and biogenesis]; tRNA ... |
215-268 |
2.10e-03 |
|
tRNA(Ile)-lysidine synthase TilS/MesJ [Translation, ribosomal structure and biogenesis]; tRNA(Ile)-lysidine synthase TilS/MesJ is part of the Pathway/BioSystem: tRNA modification
Pssm-ID: 439807 [Multi-domain] Cd Length: 235 Bit Score: 39.82 E-value: 2.10e-03
10 20 30 40 50
....*....|....*....|....*....|....*....|....*....|....*..
gi 219567478 215 DKKVICAMSGGVDSSV---AAMIVHKAVGKQLTCIFVDHGLlrKDEGDQVEDIFKKQ 268
Cdd:COG0037 15 GDRILVAVSGGKDSLAllhLLAKLRRRLGFELVAVHVDHGL--REESDEDAEFVAEL 69
|
|
| PRK13980 |
PRK13980 |
NAD synthetase; Provisional |
198-390 |
4.32e-03 |
|
NAD synthetase; Provisional
Pssm-ID: 184435 [Multi-domain] Cd Length: 265 Bit Score: 39.04 E-value: 4.32e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 198 MSSFVDEKIksikEEVGDKKVICAMSGGVDSSVAAMIVHKAVGKQLTcifvdHGLL---RKDEGDQVEDIFK--KQFNMN 272
Cdd:PRK13980 17 IVDFIREEV----EKAGAKGVVLGLSGGIDSAVVAYLAVKALGKENV-----LALLmpsSVSPPEDLEDAELvaEDLGIE 87
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 273 FIRVnaekrflqklkDISDpekkrkiIGEEFIRVFEEEAKK-LGEIAFLVQGTIYPDVVESG----LGTSAtiKSHHNVG 347
Cdd:PRK13980 88 YKVI-----------EITP-------IVDAFFSAIPDADRLrVGNIMARTRMVLLYDYANREnrlvLGTGN--KSELLLG 147
|
170 180 190 200 210
....*....|....*....|....*....|....*....|....*....|
gi 219567478 348 -----GlpeDM--DFkliEPLRELFKDEVRAVGEELGIPHKLVWRQPFPG 390
Cdd:PRK13980 148 yftkyG---DGavDL---NPIGDLYKTQVRELARHLGVPEDIIEKPPSAD 191
|
|
| lysidine_TilS_N |
TIGR02432 |
tRNA(Ile)-lysidine synthetase, N-terminal domain; The only examples in which the wobble ... |
217-315 |
6.27e-03 |
|
tRNA(Ile)-lysidine synthetase, N-terminal domain; The only examples in which the wobble position of a tRNA must discriminate between G and A of mRNA are AUA (Ile) vs. AUG (Met) and UGA (stop) vs. UGG (Trp). In all bacteria, the wobble position of the tRNA(Ile) recognizing AUA is lysidine, a lysine derivative of cytidine. This family describes a protein domain found, apparently, in all bacteria in a single copy. Eukaryotic sequences appear to be organellar. The domain archictecture of this protein family is variable; some, including characterized proteins of E. coli and B. subtilis known to be tRNA(Ile)-lysidine synthetase, include a conserved 50-residue domain that many other members lack. This protein belongs to the ATP-binding PP-loop family ( pfam01171). It appears in the literature and protein databases as TilS, YacA, and putative cell cycle protein MesJ (a misnomer). [Protein synthesis, tRNA and rRNA base modification]
Pssm-ID: 274129 [Multi-domain] Cd Length: 189 Bit Score: 38.00 E-value: 6.27e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 219567478 217 KVICAMSGGVDSSV---AAMIVHKAVGKQLTCIFVDHGlLR---KDEGDQVEDiFKKQFNMNFIRVNAEKRFLQKLKDIS 290
Cdd:TIGR02432 1 RILVAVSGGVDSMAllhLLLKLQPKIKIKLIAAHVDHG-LRpesDEEAEFVQQ-FCRKLNIPLEIKKVDVKALAKGKKKN 78
|
90 100
....*....|....*....|....*..
gi 219567478 291 DPEKKRKiigeefIR--VFEEEAKKLG 315
Cdd:TIGR02432 79 LEEAARE------ARydFFEEIAKKHG 99
|
|
| |