The first questions the new user might ask when viewing a VAST alignment in Cn3D are, "Why are some letters uppercase and others lowercase? And what are these funny '~' characters?" The short answer is that aligned residues are displayed in capital letters, unaligned in lowercase, and the '~' represents an unaligned gap. But the latter, especially, requires a bit more explanation.
Dynamic programming sequence alignment algorithms align residues based on a mutation probability score, and use gaps to model evolutionary insertions or deletions in one sequence with respect to another. An inserted residue is aligned with gap in the algorithm's bookkeeping, and is scored accordingly (a gap penalty). These aligned gaps are customarily drawn as a '-' (dash) character. This allows a continuous gapped alignment to cover a large region of two sequences, even when there are significant evolutionary differences between them.
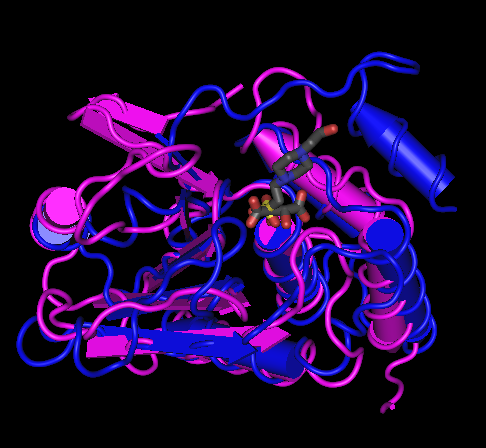
In a structure alignment (e.g. from VAST), one residue is aligned with another because their alpha carbons are nearby in space. Hence, the notion of a residue being "aligned to a gap" has no physical meaning, because an alpha carbon in one structure cannot correspond to empty space in another. Instead, the alignment is made discontinuous: aligned regions - helices or strands that overlap closely and continuously between two structures - are separated by unaligned regions, which are typically loops with sufficiently different lengths and orientations that they cannot be meaningfully superimposed.
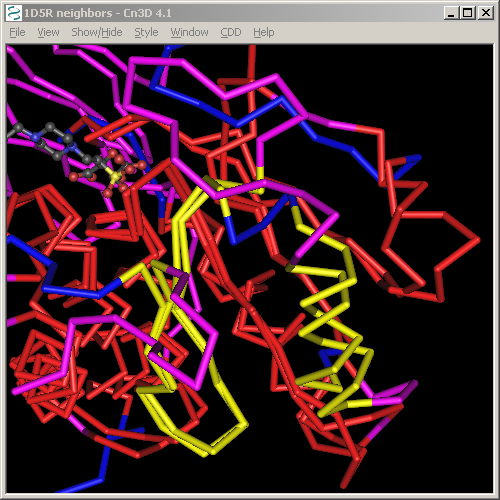
Take for example the alignment of 1D5R and 1VHR, as discussed above.

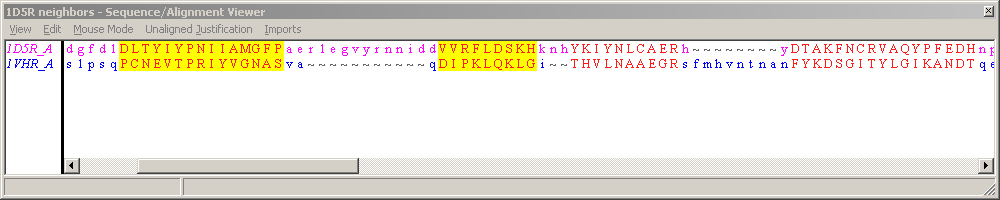
The first two blocks have been highlighted in these screen shots, showing that the first corresponds to a beta hairpin in the structure, and the second to a helix. As can be seen from the structure, these are separated by a long loop in 1D5R, but by only a short one in 1VHR.
Showing the unaligned residues inbetween the blocks, while keeping the aligned residues correctly on top of one another, requires some allowance to be made for different loop lengths. This is accomplished by using '~' characters to fill out a shorter unaligned stretch - they are gaps in the display only, and do not come from nor imply anything about the underlying alignment. Hence the shorter loop area of 1VHR is padded out with '~' characters, so that the longer loop of 1D5R has room to be displayed.
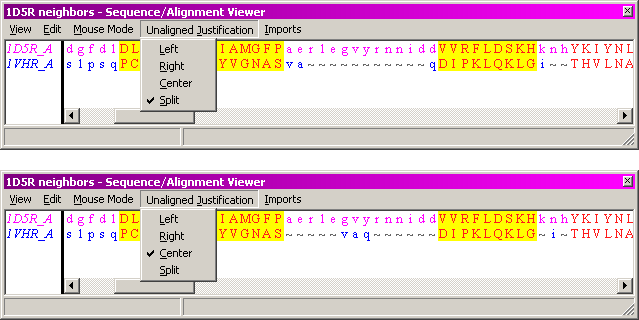
The default display for alignments has the Unaligned Justification:Split style. This shows residues from a shorter unaligned region split in the middle and put adjacent to the aligned blocks on either side. This is perfectly equivalent to the Unaligned Justification:Center option, which places unaligned residues in the center of the space between aligned blocks. These two styles are depicted below.
 ... "split" unaligned (default)
... "split" unaligned (default)
... vs. "centered" unaligned
The difference between the two is slight, but illustrates a very important point: that the unaligned residues are displayed as a convenience to the user, but nothing can nor should be inferred from the apparent "alignment" of residues in the unaligned areas (e.g., lower case letters). This contrasts directly with gapped dynamic programming alignments, where the number, length, and position of the gaps affect the score. The two displays above would score very differently if interpreted as gapped continuous alignments.
Another important issue is how Cn3D creates what looks like a multiple alignment when more than two structures are viewed, out of what is actually a series of master/slave pairs. This is discussed separately in a page about the "intersect by master" algorithm.