| |
| |
VAST, short for Vector Alignment Search Tool, is a computer algorithm developed at NCBI and used to identify similar protein 3-dimensional structures by purely geometric criteria, and to identify distant homologs that cannot be recognized by sequence comparison. The similar 3D structures identified by VAST are also referred to as "structure neighbors." Superpositions of the similar structures, and their corresponding sequence alignments, can be viewed interactively in NCBI's free Cn3D structure viewing program.
|
|
| |
Reference:
The following article describes the VAST algorithm and provides examples:
 Gibrat JF, Madej T, Bryant SH. Surprising similarities in structure comparison. Curr Opin Struct Biol. 1996 Jun; 6(3): 377-85. [PubMed] Gibrat JF, Madej T, Bryant SH. Surprising similarities in structure comparison. Curr Opin Struct Biol. 1996 Jun; 6(3): 377-85. [PubMed]
The VAST publications page provides additional references, including information about citing VAST.
Illustrated example of similar structures:
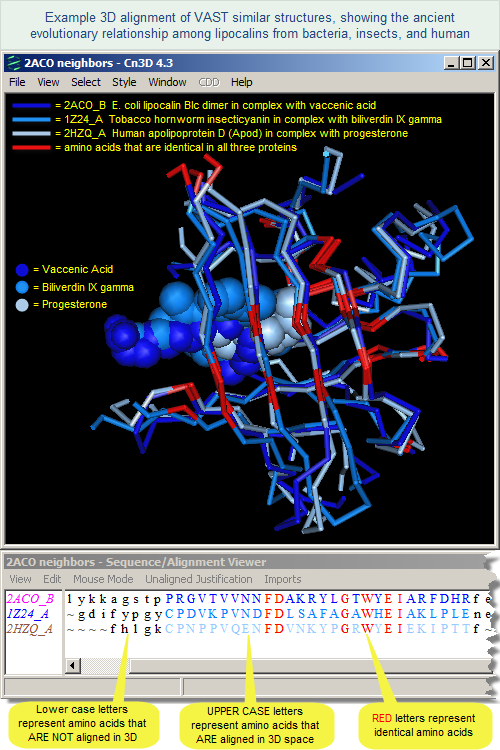
In addition to the examples provided in the reference, the illustration at the right shows a superposition of lipocalins from bacteria, insect, and human. Click on the image to open the interactive 3D alignment in the free Cn3D program. Please note that Cn3D 4.3.1 must be installed in your computer in order for the file to open. The Cn3D Tutorial provides additional details about viewing structure alignments in Cn3D.
Two versions of VAST:
The original VAST, described in this document, (1) lists the protein molecules ("chains") in the query structure, and the 3D domains that were identified in each protein, and (2) retrieves structures that are similar in shape to any individual protein molecule or 3D domain from the structure (illustrated example of original VAST results).
The newer VAST+ groups 3D-similar structures based on their degree of similarity (complete or partial) to the biological unit of the query structure, and ranks them by the number of protein molecules in the query that simultaneously match the 3D shape of protein molecules in the VAST neighbor (illustrated examples of VAST+ results).
Additional details:
The data processing:geometrical features section of the MMDB help document provides more information about how the 3D domains and similar structures are identified.
The VAST+ help document provides more information about the difference between VAST and VAST+.
| |
 |
|
|
|
| |
| |
VAST is applied on every protein in the Molecular Modeling Database (MMDB) during MMDB data processing in order to identify similar 3D structures. The pre-computed results are accessible from a structure's summary page. To retrieve them, you can use any one of the following methods:
- Enter the structure's MMDB ID or PDB ID in the "Retrieve pre-computed results" section of the VAST home page and press "GO". By default, the results will be displayed in the new VAST+ display format, which focuses on similarities between the macromolecular complexes of the query structure and hits (illustrated examples).
If you prefer to see the original-style VAST results, which focus on similarities between individual protein molecules or 3D domains within the query structure and hits, click on the "original VAST" button near the upper right corner of the VAST+ search results page. That will open a table which lists the individual protein molecules (and 3D domains, if present) in your query structure (illustrated example). Click on any link beside a protein molecule or 3D domain of interest to view a list of structures that contain a similarly shaped protein molecule or 3D domain.
— OR —
- Retrieve a structure of interest from MMDB and open its structure summary page. Follow the "Similar Structures: VAST+" link near the upper right corner of a structure's MMDB structure summary page. That will open the new VAST+ search results page (illustrated examples), which lists the query structure followed by similar structures, ranked by the degree of similarity to the query structure's biological unit.
If you prefer to see the original-style VAST results, which focus on similarities between individual protein molecules or 3D domains within the query structure and hits, click on the "original VAST" button near the upper right corner of the VAST+ search results page. That will open a table which lists the individual protein molecules (and 3D domains, if present) in your query structure (illustrated example). Click on any link beside a protein molecule or 3D domain of interest to view a list of structures that contain a similarly shaped protein molecule or 3D domain.
— OR —
- Retrieve a structure of interest from MMDB and open its structure summary page. View the "show annotation" graphic for any protein molecule of interest, then click on the bar graphic for the overall protein molecule or for any 3D domain it contains in order to view a list of other structures that are similar in shape to the molecule or 3D domain you selected.
The VAST+ help document provides details about the differences between VAST and VAST+, as well as an illustrated example of original VAST results and illustrated examples of VAST+ results.)
Note: If you have a newly resolved protein structure that is not yet in MMDB, you can use the VAST Search service to input your data in PDB file format and compare your structure against all those in MMDB. See details in the FAQ below, on "How can I compare a newly resolved 3D structure against all of the structures in the Molecular Modeling Database (MMDB)?"
|
|
|
| |
| |
| |
Whether you retrieve similar structures for a newly resolved structure, or follow the "similar structures" link in a structure record that's available in the public database, the original style VAST display will provide the following information:
- list of the protein molecules ("chains") in the query structure, and the 3D domains that were identified by the VAST algorithm in each protein
- list of the structures that are similar in shape to any individual protein molecule or 3D domain of your query structure, with links to views of their sequence alignments and 3D superpositions to the query structure.
An example of the original style VAST display is illustrated below.
- Part 1 of the illustration shows colored bars that represent the compact substructures, or 3D domains, detected by VAST in the query structure's protein molecule(s). These 3D domains serve as the fundamental unit of structure comparison. (The data processing:geometrical features section of the MMDB help document provides more information about how the 3D domains and similar structures are identified.) To view the 3D domains that have been identified in a protein molecule, open the MMDB structure summary page for a structure of interest, sroll down to the table of molecules and interactions, and click on the "show annotation" link for the protein of interest. (For example, open the structure summary page for 1PTH.)
- Part 2 of the illustration shows the original-style VAST display, which consists of a table summarizing the protein molecules and 3D domains in the query structure, and the number of structures that are geometrically similar to each individual protein molecule or 3D domain in your query structure. For any protein molecule or 3D domain of interest, click on the link in the "# of Related Structures" column to view a list of similar structures. The list includes a graphical display of alignment footprints and provides options to view sequence alignments and superposed structures.
To explore the example interactively, you can open a live web page with original-style VAST results for 1PTH, or
click on the image below to open an interactive view of the 3D alignment of 1PTH's protein A, domain 1 and a sample similar structure, 1EQG (Ovine Cox-1 Complexed With Ibuprofen). (Please note that Cn3D 4.3.1 must be installed in your computer in order for the file to open. The Cn3D Tutorial provides additional details about viewing structure alignments in Cn3D.)
The original style VAST display is still accessible by clicking the "Original VAST" button near the top of a VAST+ search results display. This document describes the features, functions, and graphics of the original style VAST display.
|
|
| |
| |
Example - Original style VAST display (as of 07 November 2013) for 1PTH, "The Structural Basis of Aspirin Activity Inferred From the Crystal Structure of Inactivated Prostaglandin H2 Synthase" (sheep prostaglandin H2 synthase)

Open a live web page with original-style VAST results for 1PTH.
|
|
|
|
| |
In contrast to the original VAST display shown above, which focuses on similarities between individual protein molecules or 3D domains, the newer VAST+ display groups 3D-similar structures based on their degree of similarity (complete or partial) to the macromolecular complex (biological unit) of the query structure; ranks them by the number of protein molecules in the query that simultaneously match the 3D shape of protein molecules in the VAST neighbor; and enables you to view the sequence alignments and 3D superpositions of the biological units. The VAST+ help document provides illustrated examples of VAST+ results and additional details about the difference between VAST and VAST+.
|
|
|
|
| |
| |
After you select a protein molecule ("chain") or 3D domain of interest from the initial VAST results page, you will see a brief list of structures that are similar in shape to the protein molecule or 3D domain you selected.
By default, the results are shown as a Graphics display (illustrated below) and list only a "medium redundancy" subset of structure neighbors, with red bars representing the alignment footprint of each structure neighbor relative to the query protein. (More details about the display are provided below the illustrated example.)
Controls near the top of the page allow you to change to a Table display (descibed in the next section of this document), and/or to increase or decrease the number of hits shown on the VAST results page with options that range from a "low redundancy" subset of proteins from structure records to "all sequences." After you select the desired options, be sure to press the "List" button in order to refresh the display.
The similar structures can be displayed as Graphics (illustrated below) or as a Table (descibed in the next section of this document).
|
|
| |

|
|
| |
The identifier for each structure neighbor is shown in the format of PDB ID + protein chain ID + 3D domain ID (e.g., 1Q4G A 4, which represents domain 4 in protein chain A from the 1Q4G structure record).
The red bars indicate the region/residues of the query domain that can be superimposed on residues from each neighbor. The gray bars and blank space are unaligned regions. These region colors are the same as those shown in Cn3D when a structure superposition is viewed in Cn3D. When the mouse is over each icon, it will display a description of what it represents.
On the sequence ruler next to the query protein ("1PTH A" in the illustration above), the aligned region indicates a sum of regions from all neighbors. This indicates the maximum fragment in the query that is similar to some other structures. The individual 3D domains in the chain are indicated by rectangles below the sequence ruler with different colors and numbers. MMDB's 3D domains are defined on the basis of structural compactness. Red indicates the query domain. Links to the conserved domain database are provided for convenience, to provide names and descriptions (where possible) of the 3D domains to which they correspond.
The check box at the left hand side of a structure neighbor's "row"
allows for selection of individual neighbors and their 3D superposition.
Clicking the sequence identifier beside it will go to the Entrez sequence page
of the neighbor. The red aligned regions in a neighbor's sequence are
displayed at the positions of their equivalent residues in the query sequence.
Clicking on these will display an HTML view of the sequence
alignment between the query and the neighbor. One of the VAST similarity
measures used for sorting (here, the alignment length: e.g., 551 residues
residues from 1PTH_A are aligned with 1Q4GA) is listed at the right hand side of the row. Clicking the name of the similarity measure (i.e., "Ali_len" in our example) will display a table with all of the VAST statistics.
|
|
|
|
| |
| |
The display controls at the top of a VAST results page allow you to change the display from the default "Graphics" format (described in the previous section) to a "Table" format.
The "Table" display lists the identifier for each structure neighbor in the format of PDB ID + protein chain ID + 3D domain ID (e.g., 1Q4G A 4, which represents domain 4 in protein chain A from the 1Q4G structure record), its description, and a number of measures of structural similarity. The columns in the table include:
- Check box: Allow you to select the structure neighbors you'd like to view in a 3D alignment with the query protein structure.
- PDB: The four-character PDB-Identifier of the structure neighbor. Click on the Identifier to switch to the MMDB Summary page of the respective neighbor.
- C: The PDB chain name. A blank space indicates that the chain does not have an identifier (many protein structures have a single chain only). Note that non-alphanumeric characters such as dashes, hyphens, underscores, etc. may be used as chain names by PDB.
- D: The MMDB 3D domain identifier. Domains are parsed based on geometrical criteria (the ratio of intradomain contacts to interdomain contacts) by an automatic method and can be visualized with Cn3D.
- Aligned Length: The number of equivalent pairs of C-alpha atoms superimposed between the two structures, i.e. how many residues have been used to calculate the 3D superposition.
- SCORE: The VAST structure-similarity score. This number is related to the number of secondary structure elements superimposed and the quality of that superposition. Higher VAST scores correlate with higher similarity.
- P-VAL: The VAST p value is a measure of the significance of the comparison, expressed as a probability. For example, if the p value is 0.001, then the odds are 1000 to 1 against seeing a match of this quality by pure chance. The p value from VAST is adjusted for the effects of multiple comparisons using the assumption that there are 500 independent and unrelated types of domains in the MMDB database. The p value shown thus corresponds to the p value for the pairwise comparison of each domain pair, divided by 500.
- RMSD: The root mean square superposition residual in Angstroms. This number is calculated after optimal superposition of two structures, as the square root of the mean square distances between equivalent C-alpha atoms. Note that the RMSD value scales with the extent of the structural alignments and that this size must be taken into consideration when using RMSD as a descriptor of overall structural similarity.
- %Id: Percent identical residues in the aligned sequence region. This is a raw measure of sequence similarity in the parts of the proteins that have been superimposed.
- LHM: Loop Hausdorff Metric. A Loop Similarity measure that shows how well two structures conform to each other in the loop regions, after structural superposition. The "loop regions" are the parts of the structures between aligned secondary structure elements (helices and strands). LHM is measured in Angstroms, with a smaller value indicative of greater similarity. The loop similarity may be undefined (indicated by 'NA') if there are too many residues with missing coordinates in the loops. Citation: Analysis of protein homology by assessing the (dis)similarity in protein loop regions
- GSP: Gapped Score. A combination (algebraic) score that uses RMSD, aligned length, and the number of gapped regions in the alignment. A smaller gapped score correlates with greater similarity. Citation: Comprehensive evaluation of protein structure alignment methods: scoring by geometric measures.
- Description: A string parsed out of PDB's COMPOUND records that describes the nature of the structure neighbor.
|
|
|
| |
| |
| |
MMDB chains are clustered into groups according to their amino acid sequence similarity in pairwise comparisons. A representative chain is selected from each group to compile a non-redundant subset of MMDB, and only one representative of each group is shown in a neighbor-list calculated by VAST. By default, a lower level of redundancy at 10e-40 is used to report structure neighbors. This keeps the table shorter while providing the most informative summary of structural relationships in MMDB.
All-against-all pairwise comparisons of MMDB-domains are calculated with the BLAST algorithm, setting a fixed database size parameter of 500,000 residues. Sequences are then clustered into groups by single linkage, whereby a sequence is merged into a group if it shows a BLAST p value of C or less with any member of the group. There are 5 levels of redundancy defined in MMDB database:
- Low redundancy: representatives are chosen from each group where sequences show a BLAST p value of 10e-7 to each other
- Medium redundancy: representatives are chosen from each group where sequences show a BLAST p value of 10e-40 to each other
- High redundancy: representatives are chosen from each group where sequences show a BLAST p value of 10e-80 to each other
- Non-identical sequence level: representatives are chosen from each group where sequences are not identical to each other
- All sequences level: this is the most redundant level, which includes all of MMDB sequences
Within each cluster of similar protein chains, cluster members are ranked
according to the apparent quality and completeness of the structure data.
The following criteria are used (ranked by decreasing priority):
- Low fraction of residues with unknown residue type
- Low fraction of residues with incomplete coordinates
- Low fraction of residues with missing coordinates
- Low fraction of residues with incomplete side-chain coordinates
- High resolution
- High number of chains (subunits) contained in the PDB entry
- High number of heterogens contained in the PDB entry
- High number of different types of heterogens.
- Chain length
For the display of structure neighbors calculated by VAST, the highest ranking chain (according to the criteria above) from each cluster found in the list of neighbors is reported. In most cases this implies that the parent structure is also similar to the other members of the sequence redundant cluster. To have them displayed, the user must select a higher level of redundancy.
|
|
|
|
| |
| |
The display controls at the top of a VAST results page allow you to change the appearance of display from Graphics to Table format. The graphic is helpful to understand the superpositions between a query domain and its neighbors. The table is good for viewing or saving the statistics from a VAST calculation.
The VAST similarity measures reported for each neighbor can be used to determine sort order. The lengths of the whole graphic and table are strongly influenced by the display subset, which determines the level of sequence redundancy chosen.
The display controls also allow you to change the number of structure neighbors listed in the display. A brief subset of structure neighbors is shown by default. You can choose increase or decrease the number of hits shown on the VAST results page by using the "List" options, which range from a "low redundancy" subset of proteins from structure records to "all sequences."
The total number of neighbors displayed in a page is limited. At most 60
neighbors from a non-redundant subset can be displayed simultaneously on one
page. In addition, by clicking check boxes to select from previously listed
neighbors, at most another 40 neighbors can also be displayed in the same
page. Therefore the maximum capacity of one page is 100 neighbors. This
feature, together with the pagination, is able to keep interesting neighbors from different pages displayed together. The page can be selected from the third pull-down menu in the "List" line.
The "Advanced similar structure search" options allow you to search for specific structures in your current set of search results. For example, if you know that a particular structure should be in your VAST results but you don't see it in the currently displayed subset of hits, you can use the "Find" button to look for that structure by MMDB, PDB, or 3D-Domain identifier. If you have done a previous search in the Entrez Structure (MMDB) database and want to find out if any of the structures retrieved by that search are in the current VAST results, you can use the "Entrez History" function in the "Advanced similar structure search" panel. That will show you the intersection, if any, of the previous Entrez Structure and current VAST search results.
|
|
|
| |
| |
| |
On a VAST results page (in either the "Graphics" or the "Table" display), individual structure neighbors can be selected by clicking in the check boxes at the left margin. Then if one chooses the button labeled "View 3D Structure", the 3D superposition of the query protein with the selected neighbors is displayed in Cn3D. Up to 10 neighbors may be viewd in a superposition simultaneously, if Cn3D without the cache mechanism is selected (this is the default). This selection also works for Cn3D version 3.0.
Although the default is to submit all atoms for display in Cn3D, the "Backbone" option can be used to control the size of the files being downloaded by Cn3D, in order to save time and memory for data transmission to the viewer. With the release of Cn3D version 4.0, the Cn3D/Cache mechanism is used to store downloaded structure data locally. With this option, the number of neighbors for display is not limited. The user must take care not to exceed the physical memory available in his/her computer. If available memory is exceeded, Cn3D will not operate properly.
The Cn3D Tutorial provides additional details about viewing structure alignments in Cn3D.
Alternatively, instead of viewing the 3D superpositions, the data can be
examined or saved to disk as a local file, for browser-independent or later
viewing. Also if the "List" "Asn1" option is selected instead of the "List"
"Graphics" or "List" "Table" from the last menu, a complete alignment file will be saved locally, including all of the neighbors in the subset.
|
|
|
| |
| |
| |
If the "View Alignment" button is chosen, a multiple alignment view will be opened in HTML, text, or FASTA with Gap formats. The check boxes at each neighbor "row" allow one to add the "Selected" neighbors into the alignments. The "All on page" option will allow a display of multiple alignments made from all of the neighbors on the same page.
The HTML- and text-format alignment views indicate aligned vs. unaligned residues as uppercase and lowercase letters, respectively. In HTML views, columns with identical residues aligned across all selected sequences are colored red, whereas those with different aligned residues are colored blue. Those not covered by all sequences will be shown in grey.
|
|
|
|
| |
| |
There are a few different reasons for this condition. One reason is simply that VAST does not consider this structure to be sufficiently similar to any other structure in the MMDB database. The VAST data use a statistical significance cutoff of P < 0.0001. This cutoff was set to be conservative intentionally, to reduce the number of false positives, but some hits that are biologically significant may be omitted because of this statistical threshold.
There are also some entries where the VAST calculation was not done: those for proteins with fewer than 3 secondary structure elements (SSEs), and structures containing no protein chains (i.e., only DNA or RNA). The molecule type and SSE count can be checked out by examining the structure with Cn3D.
|
|
|
|
|
|